He encontrado que alguna farmacia puede tener existencias limitadas de ciertos medicamentos, mientras que otras pueden tener casi cualquier formato que se le ocurra y el habitual de dosis habitualidad apareció. En resumen, siempre se contiene el almacén de corroborar. Al mismo tiempo que el producto que más que gustaba ha resultado no estaba disponible en stock otro distinto por las Buenas costumbres también debe buscarse jefe no asн parezca. Por eso es importante disponer de un Plan B para actuar cuandod ello no ocurra.
Ventaja de tomar un genérico en lugar de Asix
Un genérico es más barato que el nombre de marca
Uno de los mayores incentivos para someterse al Dónde comprar Lasix genérico en lugar de pagar la marca es que usted puede obtener un ahorrando importantes Lasix genérico. Por lo tanto, un Lasix genérico es en general mucho más barato que el homólogo de marca, así que una denominación genérica se hace posible para las personas que usan este medicamento con frecuencia. Un ejemplo: La compra de lurosemida en lugar de Lasix es una considerable ahorro para el presupuesto mensual de medicamentos.
7e1503000683.pdf
PROTEINS: Structure, Function, and Genetics 53:683– 692 (2003)
Automatic Annotation of Protein Function Based on Family Identification Federico Abascal* and Alfonso Valencia Protein Design Group, National Centre for Biotechnology, CNB-CSIC, Cantoblanco, Madrid, Spain ABSTRACT Although genomes are being se- accessed at http://www.pdg.cnb.uam.es/funcut.html. quenced at an impressive rate, the information The programs are available upon request, although generated tells us little about protein function, installation in other systems may be complicated. which is slow to characterize by traditional meth- Proteins 2003;53:683– 692. 2003 Wiley-Liss, Inc. ods. Automatic protein function annotation based on computational methods has alleviated this imbal- Key words: protein function prediction; genome ance. The most powerful current approach for infer- analysis; protein families and subfami- ring the function of new proteins is by studying the lies; orthologues and paralogues; anno- annotations of their homologues, since their com- tation inconsistencies; database errors mon origin is assumed to be reflected in their struc- ture and function. Unfortunately, as proteins evolve INTRODUCTION they acquire new functions, so annotation based on
Spectacular progress has been made in the automation
homology must be carried out in the context of
of experimental techniques in molecular biology, espe-
orthologues or subfamilies. Evolution adds new com-
cially those for genome sequencing, functional genomics,
plications through domain shuffling: homology (or
and proteomics. In all cases, however, it is difficult to reach
orthology) frequently corresponds to domains rather
valid biological conclusions based on the massive data
than complete proteins. Moreover, the function of a
generated by such approaches and the development of
protein may be seen as the result of combining the
computational methods is still a bottleneck. The two key
functions of its domains. Additionally, automatic
steps in the analysis of genomic data are the identification
annotation has to deal with problems related to the
of the genes in the raw DNA sequence and the prediction of
annotations in the databases: errors (which are
the function of the corresponding open reading frames
likely to be propagated), inconsistencies, or differ-
(ORFs). This study focuses on the second step. ent degrees of function specification. We describe a method that addresses these difficulties for the THE PROCESS OF FUNCTION PREDICTION BY annotation of protein function. Sequence relation- TRANSFERENCE OF INFORMATION FROM ships are detected and measured to obtain a map of HOMOLOGOUS SEQUENCES the sequence space, which is searched for differenti-
The first step is the search for similar sequences in
ated groups of proteins (similar to islands on the
databases, followed by the selection of homologous se-
map), which are expected to have a common func-
quences from the set of similarities, i.e., identifying se-
tion and correspond to groups of orthologues or
quences with a common evolutionary origin. The statistics
subfamilies. This mapmaking is done by applying a
of sequence similarities1 are commonly used for this
clustering algorithm based on Normalized cuts in
purpose. Unfortunately, identification of the homologues
graphs. The domain problem is addressed in a simple
is not sufficient to guarantee a correct transference of
way: pairwise local alignments are analyzed to deter-
functional annotation.2–6 In the course of evolution, homo-
mine the extent to which they cover the entire
logues differentiate to embrace new functions, correspond-
sequence lengths of the two proteins. This analysis
ing to the organization of sequence families and subfami-
determines both what homologues are preferred for
lies.7 For example, the superfamily “P-loop containing
functional inheritance and the level of confidence of
nucleotide triphosphate hydrolases” includes families as
the annotation. To alleviate the problems associated
varied as “RNA helicases,” “G proteins,” and “ABC trans-
with database annotations, the information on all
porters.” Moreover, in the “G proteins,” families special-
the homologues that are grouped together with the query protein are taken into account to select the most representative functional descriptors. This
Grant sponsor: Spanish Ministry of Science and Technology (CICYT);
method has been applied for the annotation of the
Grant sponsor: Government of Madrid. genome of Buchnera aphidicola (specific host
*Correspondence to: Federico Abascal, Protein Design Group, Na-
tional Centre for Biotechnology, CNB-CSIC, Cantoblanco, Madrid
Baizongia pistaciae). Human inspection of the anno-
E-28049, Spain. E-mail: [email protected]tations allowed an estimation of accuracy of 94%;
Received 14 January 2003; Accepted 25 February 2003
the different kinds of error that may appear when using this approach are described. Results can be
ized in various cell functions co-exist, such as ras-related
tions. It is quite common to find annotations of different
proteins, involved in the cell cycle; rab, related to vesicle
natures or ones addressing a different level of detail for a
cell traffic; arf, also part of the protein trafficking machin-
given function. For example, of two proteins belonging to
ery; elongation factors Tu and G, and others. The process
the ras-p21 subfamily, Q9BJ57 is annotated as “Ras” in
of transference of functional annotation based on the
TrEMBL, while RASH_HUMAN is annotated as “Trans-
identification of homologues requires a deeper analysis of
forming protein P21/H-RAS-1 (C-H-RAS)” in Swiss-Prot,
the structure of the corresponding families and their
with a deeper definition of the protein function. An ex-
ample of different degrees of specificity may be found in
A complete reconstruction of the phylogenetic tree of the
the case of PS2_HUMAN and PS2_MOUSE, annotated as
family could be required for the analysis of the relation
“PS2 protein precursor (HP1.A) (Breast cancer estrogen-
between family groups and functions. Unfortunately, the
inducible protein) (PNR-2)” and “PS2 protein precursor,”
process of tree building and family analysis is difficult to
respectively. In the first case, given the importance of the
data that relate this protein to human cancer, this specificinformation is included in the description of the function. Function-Prediction Errors Introduced by
Therefore, the analysis of the annotations requires a
“Classical” Annotation Strategies
balance between general descriptions and detailed ones,
Most of the current systems for automatic annotation
corresponding to higher accuracy but less information in
obviate the step of analyzing homologies in terms of
families and subfamilies and directly transfer functionfrom the most similar sequence identified in the data-
RELATED APPROACHES TO THE AUTOMATIC
bases, for example, by selecting the best hit after searching
PREDICTION OF PROTEIN FUNCTION BASED
with standard tools such as BLAST.8 The obvious diffi-
ON THE INFORMATION OF HOMOLOGOUS
culty of encapsulating in a single relation all the functional
SEQUENCES
information associated with a given protein family makes
Some of the recent approaches are introduced in the
this annotation process a poor substitute for the deeper
analysis of the structure of the protein family structure.
GeneQuiz13 was the first system that completely auto-
There is copious literature on the errors introduced by the
mated the task of sequence analysis and function annota-
iteration of this process (see for example Brenner9 and
tion. The functional information was obtained from the
Devos and Valencia10). Indeed, the systematic comparison
potentially most informative sequence selected among the
of the annotations stored in databases has made it clear
set of similar ones. Rules for the selection of informative
that a significant number of homologous sequences have
sequences were related to the database of origin, presence
of fragments, and type of keywords associated to them.
The correct characterization of the relation between the
Genequiz delivered annotations with an associated confi-
sequences forming a protein family may solve some of
dence value that was related with the similarity (fixed
these problems, even if the final process of annotation still
e-value) with the sequence selected for the information
relies on the existing database annotations. Hence, such
transfer. The system included a simplified lexical analysis
characterization could, therefore, be subject to the errors
for identifying the informative descriptions (it has inspired
introduced during this process. Efforts are underway to
the lexical analysis that we apply).
improve the database annotations by keeping pointers to
EditToTrembl14 is a system in which the annotation of
the origin of the annotations. An attempt is also being
sequences is based on the intricate execution of different
made to create systems that facilitate this process by
programs to predict such features of the proteins as
automatically retrieving information from the original
transmembrane regions, subcellular location, or enzy-
matic codes. A complement of this work is that of Fleisch-mann et al.,15 where a method for automatic functional
Domain Shuffling and Other Difficulties
annotation is described. Thus, these authors overcome
The process of classifying protein families into the
some of the previously mentioned difficulties, such as the
corresponding families and subfamilies is not trivial. The
functional transfer from the best hit. The method is based
study of the evolutionary relationships between proteins is
on the use of PROSITE16 as an external database to
often complicated by the presence of multiple domains.
cluster the sequences of a reference database (Swiss-
Domains can have different origins, and be associated with
Prot17 in this case) into groups. Each group is inspected to
several domains in different proteins. Moreover, the func-
identify functional information shared by most of the
tion of a protein can be seen as the result of combining the
proteins. In the positive cases, an annotation rule is
functions of its domains. Therefore, for any function
derived for the annotation of new sequences. Additionally,
prediction schema it is essential to take into account the
some rules are applied to reduce the potential number of
domain structure of proteins and avoid transferences
false positives, for example, the taxonomy of the query
based on incomplete domain identification.
sequence must match the species distribution of the pro-
After identifying the sequence relations in the corre-
teins described by the (PROSITE) condition. This conserva-
sponding families, however, further difficulties arise asso-
tive approach is currently applied during the generation of
ciated with the transference of the corresponding annota-
the TrEMBL entries, producing an enrichment of the
functional annotations prior to the more precise annota-
in different categories depending on the extent to which
tion work carried out by the Swiss-Prot curators. Using the
the alignments cover the length of the query and target
PROSITE groups as seeds of the process imposes limita-
sequences (alignment categories).
tions for the coverage of the annotations.
3. Key functional annotations of the corresponding pro-
The PRECIS system18 is more a distiller of information
teins are analyzed, including functional descriptions,
than an annotation method. Basically, it receives a set of
enzymatic activity codes, and Swiss-Prot style key-
Swiss-Prot identifiers (which could come, for example,
from a BLAST report) and distils a functional report
4. The transference of information is carried out starting
combining their annotations, thus removing redundancies
from the alignment categories with a better coverage. A
and applying some rules and filters for the different fields
confidence level is assigned to each one of the annota-
tions. This level is derived from the alignment catego-
Andrade6 described a method for addressing the prob-
lem of the annotations specific to protein domains by usingposition-specific annotation of protein function based on
Step 1. Sequence searches
the analysis of multiple homologous sequences. The mul-
The similarity searches were carried out with BLAST on
tiple sequence alignment corresponding to the homologues
a non-redundant database (nrdb program from NCBI at
was used to assign functions to specific domains (positions
ftp://ftp.ncbi.nih.gov/pub/nrdb and cd-hit19) that included
covered by the sequences in the alignment). The functional
Swiss-Prot, TrEMBL, and TrEMBLnew. Those sequences
descriptions of the similar sequences were processed and
with a similarity value above a cut-off (e.g., E-value Ͻ 0.1)
screened for common strings of words. The correlation
were further BLAST-aligned between them to obtain a
between the conservation of the aligned positions with
rough measure of their pairwise similarity (i.e., their
respect to the query protein and the presence of common
E-values). This procedure effectively maps the sequence
functional descriptors in the aligned proteins was used to
space surrounding a query sequence. If desired, this local
produce annotations common to the shared positions. This
sequence space map can be extended to other more diver-
procedure allowed the construction of consensus descrip-
gent related subfamilies through iterative intermediate
tions attached to defined regions of the query protein. The
sequence searches,20,21 exploiting the transitivity prin-
two main difficulties of this idea are perhaps the complex-
ciple of homology: if protein A is homologous to B, and B to
ity of its automation and the fact that it will work properly
C, then A and C are homologues (if the domains shared in
only under certain conditions (for example, it requires that
the A-B and B-C relationships correspond to each other).
the set of homologues contain proteins belonging to the
Moreover, recursive searches provide better descriptions
of the sequence space, so the clustering works better. The
The approach presented here tries to overcome many of
use of PSI-BLAST or other profile-based methods is inad-
the aforementioned obstacles for function annotation. It
equate for this classification process because PSI-BLAST
applies a clustering algorithm to classify proteins into
does not return measures of the distance between pairs of
families and subfamilies. A lexical analysis inspired on
proteins but distance between proteins and profiles. A
GeneQuiz is applied to identify informative functional
post-processing of the PSI-BLAST results by, for example,
descriptions. As in Andrade6 and Fleischmann et al.,15 our
realigning all the results, could be a valid alternative.
approach uses information from multiple homologues to
Identification of subfamilies by clustering. The com-
select the information to be transferred. The problem of
plete set of distances between the sequences provides the
transferring functions from unrelated domains is ad-
basic description of the local sequence space. A clustering
dressed by analyzing the degree of coverage of the corre-
process is used to identify groups of sequences that more
sponding local sequences alignments.
likely correspond to protein subfamilies. The algorithmused is the “Normalized Cut,”22 and the application of this
graph theory to biological sequences is described in Abas-
Algorithm for the Assignment of Functional
cal and Valencia.23 A weighted undirected graph G(V, E)
Annotations Based on the Analysis of Sequence
represents the sequence space. The nodes (V, sequences in
Clusters
this case) are connected through arcs (E) that represent
The workflow of the method proceeds as follows:
their similarity relationship. Each arc has an associatedweight (w), proportional to the similarity measured be-tween the sequences in the form of Ϫlog (E-value) of the
1. A sequence similarity search is carried out to find
BLAST algorithm. A cut (A, B) in a graph is a partition of G
proteins related to the query sequence. A clustering
into two sets of nodes A and B, obtained by removing some
algorithm is applied in order to identify closely related
of the arcs. The capacity of a cut is the sum of the weights
sequence groups in the set of similar proteins. More
of the arcs that have to be removed to obtain the cut (A,B).
related sequences are more likely to share a common
The minimum cut24 of a graph is the one with minimum
function. In some cases, recursive sequence similarity
capacity. The minimum cut provides an effective measure
searches lead to better representation of the related
of the separation of the initial sequence space in two
subfamilies, which facilitates the clustering.
2. The local alignments with the closely related proteins
clustered together with the query protein are classified
Shi and Malik22 proposed a normalization of the capac-
provided in the standard “DE” field of Swiss-Prot. Having
ity of the cuts by including in the formula the amount of
a set of proteins belonging to the same subfamily (or group
connections of each one of the two separate sequence
of orthologues) facilitates the secure transfer of a good
functional description, since we can select the most repre-sentative description (the most homogeneous compared to
Ncut(A,B) ϭ cut(A,B)/asso(A,V) ϩ cut(A,B)/asso(B,V)
where asso(A,V) is the sum of the weights of the arcs fromall nodes in A to all nodes in V (including those in A).
1. Noninformative word filtering. Descriptions are filtered
Once the best cut is determined, the algorithm proceeds
to remove words that contain no information about
recursively, searching for new cuts of the established
function, such as FRAGMENT, HYPOTHETICAL, or
sequence groups. The recursive process continues until
2. Deriving a “homogeneity” score for each description tomeasure how representative it is. Each description is
1. The arithmetical mean of the arc weights in A or B
split into its component words, and for each word the
exceeds by twofold the arithmetical mean of the arc
frequency with which these words appear in the set of
descriptions in the cluster is calculated. A pre-score is
2. The number of arcs divided by the maximal possible
calculated for each description by adding together the
number of arcs is higher in A or B than the same
frequencies of its words. This pre-score is divided by a
correction factor to avoid the bias towards long descrip-tions. This factor is defined as the number of words
An evaluation of this clustering method applied to
divided by the number of synonyms (that usually are
protein sequences was carried out in Abascal and Valen-
very informative and are given between parenthesis in
cia.23 The outcomes of the clustering process are a number
the Swiss-Prot entries). A normalized homogeneity
of groups of well-defined and highly connected sequences
score is calculated as the fraction that each description
and the distances between these clusters. One of these
score represents in the sum of all the description scores.
groups is the one containing the query protein and ideally
3. Weighting normalized homogeneity scores with normal-
will contain the other members of its subfamily. ized similarity scores. Similarity scores are normalizedin the same manner, by calculating the fraction each
Step 2. Analysis of the Local Alignments and
BLAST similarity score represents in the total sum of
Assignment of “Alignment Categories”
BLAST similarity scores. Both normalized scores are
The local alignments of the query sequence versus the
weighted in a combined score, what is useful for the
closely related proteins are analyzed to determine the
cases where two or more subfamilies erroneously get
degree to which they cover the corresponding sequence
lengths. We have defined four categories:
As will be explained later, alignment categories and
1. Both the query and the target align through more than
these combined description scores are used to select the
80% of their lengths. In this case, the functional trans-
annotation to be transferred. Once selected, the descrip-
fer is considered to be secure and complete.
tion is inspected to reject descriptions that contain no
2. The entire length of the query sequence cannot be
information. The process of identification of non-informa-
aligned with the target sequence. In this case, the
tive descriptions is based on lexical analysis, inspired in
transference of functional annotations might not be
complete since the query protein may contain differen-
Examples of frequent non-informative annotations are
tial properties associated to the region of the sequence
“[Hypothetical|Putative] [Mol.Weight] [Lipo|Glyco]Protein
[word]” where, if “word” is present, it should be present in at
3. Less than the entire length of the target protein is
least some of the other descriptions of the proteins in the
aligned with the query protein. In this case, the transfer-
cluster to avoid rejection.“[Mol.Weight]” represents in perl
ence of annotations could be wrong if some of the
گdϩ(گ.)*(گd)*(گs)*K(D)*(A)*(گs)*. The character “|” means “or.”
functions of the target protein are associated to the
Words inside “[ ]” may or may not appear, etc.
sequence region not aligned with the query sequence.
Some of the words commonly found in non-informative
4. The less confident category corresponds to the cases in
descriptions are: “Intergenic”, “Cosmid” or “Genomic se-
which neither the query nor the target align completely.
quence,” their presence is enough to label descriptions asnon-informative. Another rule to identify uninformative
In the cases where the target protein is annotated as
descriptions is to remove all (expected) uninformative
FRAGMENT in the database, its alignment is always
words and check for the remaining words if they are
present in at least some of the others descriptions in thecluster. Step 3. Definition of the Functional Annotations to
Finally, informative descriptions are cleaned by remov-
Be Transferred
ing words that frequently appear in functional descrip-
The goal of functional descriptions is to find descriptions
tions but are not transferable based on homology, such as
of function that can be compared with the information
the molecular weight or the word “fragment.”
Key words accepted: A B C. Note that C, E, and F have the same frequency, but only C is transferred
to avoid mixing key words that do not co-occur. The process: first A is selected as seed. Then, since B isconnected to A more than 4 times (8/2), B will also be accepted. Then C will also be accepted because it isconnected to B more than 2.5 times (5/2). No more keywords will be added. Enzymatic codes. Since EC numbers are expressed in
● For each protein description, uninformative words are
a non-ambiguous language, there is no need to measure
removed and the frequency of the remaining ones is
their homogeneity. The EC number is transferred to the
query protein from the sequence inside its sequence clus-
● We build a graph in which there is one node for each of
ter with the highest similarity score and a better align-
the words and the (weighted) arcs between the nodes
reflect the number of descriptions in which two words
Key words. Functional key words assigned in Swiss-
Prot depend strongly on the functional domain organiza-
● The most frequent word is selected as seed, and the
tion of the proteins, e.g., Myristate, Calcium-binding. We
graph is searched for all those nodes (words) connected
have preferred to transfer only key words in the cases
to its node with a frequency at least half of the seed
where the target protein is completely covered by the
frequency (slightly different from the approach used
alignment (first and second alignment categories). The key
words frequency is calculated, and a graph is built using
● Then, for each of those accepted words, the most fre-
key words as nodes, and the arcs connecting the nodes are
quent position where they appear relative to the seed
labelled by the number of times that key words appearassociated to the same protein. The selection process is
based on the co-occurrence of the key words, and it is
● Finally, the description is built by sorting the list of
applied reiteratively to rescue partial co-occurrences. First,
words according to these most frequent positions. Even
a key word score is calculated for each protein by weight-
if this sorting procedure is not perfect, it is simple
ing similarity and homogeneity scores, as in the case of the
enough to give an idea of the functions that are present
functional descriptions). The key word with the highest
frequency selected among the ones of the protein with thehighest key word score is accepted, and selected as seed. Step 4. Assignment of the Functional Annotations to
Repeated searches recover other key words connected to
the Different Alignment Categories
some of the accepted ones with at least the half of itsfrequency. The frequency must be half or greater to avoid
For the final transfer of functional description, the
including key words that are not co-occurring, as illus-
proteins are inspected from the best alignment coverage
category to the worst. In each of the categories, the best
Neighbor clusters annotation. We have incorporated
description (higher combined score) is searched for. If
an additional procedure to extract information from neigh-
there are no descriptions in that category, or if the best one
boring sequence clusters. In this case, the intention is to
is considered noninformative, we go down to the following
provide general annotations for each of the sequence
category and search again for the best description in this
clusters. This procedure is particularly helpful in those
category. The confidence of the transference is derived
cases where the cluster of sequences around the query
protein does not contain enough proteins with relevantfunctional annotations.
Essentially, the procedure works by selecting the word
Selected Examples of Functional Annotation
that is most frequent in the set of descriptions and all theother words that are frequently associated to it. The
The parameters used for the recursive sequence similar-
position of these words with respect to the most frequent
ity searches have been selected to obtain clearer results in
one is used to order them and to build the final functional
each of the examples. They are different in each case
cluster description. In detail, the steps are:
because of the different sizes of the different familiesanalyzed. For the case of Buchnera’s genome annotation,
● For each of the neighbor clusters its own set of annota-
we did not use recursive searches but single BLAST
searches with realignment of all the results (see Methods). TABLE I. BLAST’s Best Hits for Swiss: TETM_NEIME†
Sequences producing significant alignments:
TET1_ENTFA (Q47810) Tetracycline resistance protein tetM from tr. . .
TETS_LACLA (Q48712) Tetracycline resistance protein tetS (Tet(S)).
TETO_CAMCO (P23835) Tetracycline resistance protein tetO (Tet (O)).
TETW_BUTFI (O52836) Tetracycline resistance protein tetW (Tet(W)).
Q93K56 (Q93K56) Tetracycline resistance protein.
TETP_CLOPE (Q46306) Tetracycline resistance protein tetP (Tetb(P)).
Q97J38 (Q97J38) Tetracycline resistance protein, tetQ family, GT.
TETM_STRLI (Q02652) Tetracycline resistance protein tetM.
OTRA_STRRM (Q55002) Oxytetracycline resistance protein.
Q97KR3 (Q97KR3) Tetracycline resistance protein tetP, contain GT.
Q8XLR6 (Q8XLR6) Probable tetracycline resistant protein.
EFG_THETH (P13551) Elongation factor G (EF-G).
Q9AIG7 (Q9AIG7) Elongation factor G.
EFG_AQUAE (O66428) Elongation factor G (EF-G).
EFG_THEMA (P38525) Elongation factor G (EF-G).
Q8YP62 (Q8YP62) Translation elongation factor EF-G.
Q9PI16 (Q9PI16) Elongation factor G.
EFG_CHLMU (Q9PJV6) Elongation factor G (EF-G).
BAB56709 (BAB56709) Translational elongation factor G.
Q9F4B2 (Q9F4B2) Translational elongation factor G, EF-G (Fragment).
EFG_SYNP6 (P18667) Elongation factor G (EF-G).
Q9RXK5 (Q9RXK5) ELONGATION FACTOR G.
†It can be appreciated that BLAST e-values order appropriately the sequences of the tet and EF-Gsubfamilies. Even if there’s not a clear separation attending to the magnitude of the e-values, the clusteringalgorithm distinguishes both subfamilies, but fails to include two more divergent tet’s in the proper cluster. Proteins TET1_ENTFA to OTRA_STRRM belong to the same group; proteins Q97KR3 and Q8XLR6 formanother cluster; the remaining ones form the third group. The complete BLAST result can be obtained from:http://www.pdg.cnb.uam.es/fabascal/SEARCH AND CLUS/TETM NEIME/Q51238.bls. TABLE II. Subfamilies Found by the Recursive Searches for Swiss::TETM_NEIME and the Subsequent Clustering, Which Resulted in 21 Clusters
Tetracycline resistance protein tet[W M S R . . .]
Peptide chain release factor 3 (RF-3) (bacteria)
Elongation factor 1-alpha plus 18 Eukaryotic peptide chain release factor 3
NodQ bifunctional enzyme and CysN/cysC bifunctional enzyme
Selenocysteine-specific elongation factor
Those containing more than 2 sequences are represented. Note that some subfamilies may be incompletebecause similarity searches were limited to a maximum of 750. TETM_NEIME
applying three rounds of intermediate sequence searches
The first selected example corresponds to the tetracy-
with a cut-off E-value of 1e-07), allowed the appropriate
cline resistance protein, (tetM) from Neisseria meningiti-
separation of the two subfamilies (Table II) and also the
dis. The BLAST search with this protein vs. a nonredun-
correct classification of other more distant subfamilies.
dant database selected at a 90% identity level rendered the
Assuming that the co-clustered sequences share a common
results shown in Table I. In this case, there is not a clear
function makes it possible to use them as sources of
separation of the subfamilies of TET and EF-G proteins
annotation, analyzing the descriptions as described in
based on E-values. However, the clustering of the se-
Methods. This yields the annotation for the query protein:
quence space local to the query protein (obtained by
(TET(S)) instead of TETM. This is an especially problem-atic case where the clustering is not able to classify intoseparate groups the different tetracycline resistance deter-minants. Instead, it puts them together according to theirhigh similarity. The annotations in the database (or thenomenclature) seem to be inconsistent (or the specificityhas no evolutionary foundation), because the percentage ofsequence identity is much higher between some Tet(M) andTet(whatever) than between two Tet(M), for example, TETM-_NEIME vs. TETS_LACLA (77%) and TETM_NEIME vs. TETM_STRLI (35%). A phylogenetic tree of the best BLASThits of TETM_NEIME can be see at http://www.pdg.cnb.ua-m.es/funcut.html. The keywords for the co-clustered se-quences were:
Q02652 Protein biosynthesis; Antibiotic resistance; GTP-
Q93K56 GTP-binding. Q46306 Protein biosynthesis; Antibiotic resistance; GTP-
Note that some subfamilies may be incomplete because
Q51238 Protein biosynthesis; Antibiotic resistance; GTP-
recursive searches were stopped before convergence. Each circle and itsradius correspond with a cluster and its size. The numbers inside indicate
the cluster id and the number of proteins in it. The width of the lines
Q47810 Protein biosynthesis; Antibiotic resistance; GTP-
connecting the clusters represents the strength of their connection. The
different gray intensities correspond to the different families.
P23835 Protein biosynthesis; Antibiotic resistance; GTP-
EC 2.7.7.4 ADENYLATE TRANSFERASE SAT ATP
O52836 Protein biosynthesis; Antibiotic resistance; GTP-
ID:18; SIZE:11; PROXIMITY:1.87 SELENOCYSTEINESPECIFIC ELONGATION FACTOR SELB TRANS-
Q48712 Protein biosynthesis; Antibiotic resistance; GTP-
These automatic annotations are difficult to read in
some cases because of the absence of punctuation charac-
Q55002 Protein biosynthesis; Antibiotic resistance; GTP-
ters, such as, for example, the parentheses, which are not
managed. Moreover, when a word appears more than once
in a description, only the first is counted. For example, the
from which were derived the following key word annota-
cluster annotation SELENOCYSTEINE SPECIFICELONGATION FACTOR SELB TRANSLATION should
tions: GTP-binding; Protein biosynthesis; Antibiotic resis-
The neighbor clusters and their annotations were:
One selenocysteine-specific elongation factor is sepa-
rated from no. 18 to the singleton cluster no. 20. If this
ID:10; SIZE:80; PROXIMITY:46.95 ELONGATION
“solitary” protein were the query protein, then annotation
would not be possible. This represents an example of what
ID:14; SIZE:24; PROXIMITY:26.95 PEPTIDE CHAIN
kind of errors may appear. Results can be accessed at:
http://www.pdg.cnb.uam.es/funcut.html.
ID:4; SIZE:35; PROXIMITY:20.42 GTP BINDINGPDXK_SHEEP
ID:3; SIZE:50; PROXIMITY:16.25 GTP BINDINGhttp://www.pdg.cnb.uam.es/funcut.html
This protein is a PYRIDOXINE KINASE. Recursively
ID:5; SIZE:74; PROXIMITY:13.45 ELONGATION
searching with it (three rounds with and Evalue cut-off of
1e-03, in this case vs. a 100% non-redundant database
ID:21; SIZE:117; PROXIMITY:10.51 ELONGATION
comprising Swiss-Prot, TrEMBL and TrEMBLnew), and
subsequently clustering the results, yields 29 clusters,
ID:2; SIZE:59; PROXIMITY:6.13 TRANSLATION
corresponding to 160 sequences (first round: 1 sequence;
second: 70; third: 89). Of these 29 groups, 7 contain more
ID:15; SIZE:248; PROXIMITY:3.07 ELONGATION
than three sequences (Fig. 2); the rest correspond to
out-layers or cases where the clustering fails to keep the
ID:17; SIZE:25; PROXIMITY:1.96 SULFATE ADENYLYL-
proteins in their corresponding families (a phylogenetic
Annotation. The subfamily of the PDXK_SHEEP is TABLE III. Buchnera aphidicola Automatic Protein
separated in an only eukarya cluster, together with 19
Function Annotation
relatives. The bacteria Pyridoxal/pyridoxine/pyridoxam-
ine kinases (including PDXY and PDXK) are divided inthree next neighbor clusters. The query protein was anno-
tated with the highest level of confidence as PYRIDOXINE
KINASE (PYRIDOXAL KINASE), with enzymatic
activity 2.7.1.35 and key words Kinase and Transferase.
The other key word in the Swiss-Prot entry corresponding
to PDXK_SHEEP, “Acetylation,” was not transferred be-cause PDXK_SHEEP was the only protein in the clusterwith that key word assigned to it.
separately from their homologues, in singletons, so nofunctional annotation transfer can be carried out. In
ID:13; SIZE:9; PROXIMITY:19.35 PYRIDOXAMINE
Tamas et al. 2002,26 this divergence for flagellar pro-
teins is also observed and it is proposed that it may be
ID:11; SIZE:4; PROXIMITY:16.63 KINASE
related to the acquisition of new functionalities, since
ID:14; SIZE:5; PROXIMITY:13.78 PYRIDOXINE
flagelles have not been observed in this bacteria. KINASE EC 2.7.1.35 PYRIDOXAL VITAMIN B6
2. Too specific descriptions (9 cases): in this case, some
specie-specific (not transferable) word in the descrip-
ID:3; SIZE:62; PROXIMITY:1.13 PHOSPHOMETH-
tion is transferred. For example, for ycfC, the automatic
annotation “Hypothetical protein ycfC (ORF-23)” was
ID:24; SIZE:22; PROXIMITY:0.69 RIBOKINASE
manually corrected to “Hypothetical protein ycfC”. Theword “ORF-23” is particular for the source specie.
With other more permissive searching parameters (e.g.,
3. Incorrect functional assignments (2 cases): Protein hscA
higher e-value cut-off and additional rounds of sequence
corresponds to “chaperone protein hscA homologue” but
searches), other remotely related subfamilies were identi-
it was automatically annotated as “chaperone protein
fied, such as: tagatose-6-phosphate kinase, phosphofruc-
dnaK” because the clustering did not separate these two
tokinases, 2-dehydro-3-deoxygluconokinase, guanosine ki-
very close subfamilies and in the cluster dnaK proteins
nase, adenosine kinase, etc. The analysis of those results
involving more clusters and isolated sequences (single-tons) was not followed here.
An illustrative example of the usefulness of analyzing
Application to the Annotation of the Buchnera
whether or not the alignments cover the entire sequence
length of the involved proteins is the one of polA. The bestBLAST hits for this protein are DNA POLYMERASES I.
This annotation method has been applied for the analy-
In the BLAST similarity list there are also proteins
sis of the genome of Buchnera aphidicola (specific host
annotated as “Probable 5Ј-3Ј exonucleases.” This buchnera
Baizongia pistaciae).25 In this case, the sequence space
protein has lost most of its domains, so alignments with
local to each of the 507 coding genes of buchnera was built
DNA POLYMERASE targets cover 90 –30% of query and
with single BLAST searches (e-value cut-off: 0.1) and all
target lengths. However, alignment with less similar
vs. all pairwise alignment of the results. The resulting
5Ј33Ј exonuclease covers 91–97%. This annotation, which
automatic annotations are available at the funcut web site.
seems to be the correct one, was detected automatically.
A comparison of automatic annotation vs. BLAST best hitannotations is also available at the same site. Keywords and EC Numbers
Annotations were manually analyzed in the cases in
For the 507 proteins in buchnera’s genome, there were
which no automatic annotation was produced and in the
281 EC number assignments, corresponding to 269 pro-
cases of conflict with the annotation of other closely related
teins (some proteins have more than one enzymatic activity).
genomes in the database. This allowed us to obtain an
In the case of key words, 1,463 were assigned to 470
approximate measure of the accuracy of this approach (see
proteins, but if we discount the frequent (but not adequate
Table III).The accuracy was estimated at 94% and three
for transfer) key word “Complete proteome,” we have 1,071
kinds of errors were established for the remaining 6%.
key words for 391 proteins. Rejection of nontransferable
Basically, these errors come from unsatisfactory cluster-
ing that in some cases divides a given subfamily. This thencreates singletons, and in other cases fails to separate two
DISCUSSION
(or more) subfamilies. Other “errors” are due to special
We have presented a method for the generation of
characteristics of the lifestyle of this obligate endo-
functional annotations based on the study of the annota-
tions of homologous sequences. The method includes newfeatures related to the specific identification of protein
1. Singleton errors (21 cases): fliH, fliJ, fliK, fliM, flgB,
subfamilies (orthologous groups) because at this level the
flgM (flagellar proteins). These proteins are clustered
function of the homologous proteins tends to be more
conserved than in general protein families (mixture of
those of other genes of known function. Function can also
paralogous and orthologous sequences).
be predicted by exploring the set of interactions deduced
Our method seems to produce correct annotations includ-
by different experimental or computational techniques (for
ing those of the “DE” and “KW” fields of Swiss-Prot and
a review see, Valencia and Pazos28). According to this idea,
enzyme classification numbers (Enzyme Commission code,
different attempts have been made to use the genomic
EC). It is obvious that these three features do not account
context to improve annotations, for example, by increasing
for all the possibilities of protein function description, and
the probability of associating a function with a given
other database annotations are also important for a com-
protein if it were the best candidate in a given genome.3,29
Even if interesting attempts to unify the various sources of
It is important to keep in mind that a description of
information on association between proteins and genes are
protein function can be done at very different levels from
underway (for an early study see Marcotte et al.30),
biochemical to cellular. It is appealing to think that the
problems of consistency and accuracy still persist, and the
levels more directly related to the chemical function will
current knowledge about pathways and networks is still
tend to be more conserved at large sequence distance.
insufficient to allow a systematic approach.
Cellular functions, then, which are more dependent on the
Another completely different path has been opened by
cellular context and interactions, will be less conserved at
the use of sequence features, (e.g., sequence length, poten-
the sequence level. Part of this complexity in function
tial phosphorylation sites, predicted TM segments), for the
description is quite obvious in the comment (“CC”) and
prediction of protein functional class.31 It is conceivable
feature (“FT”) Swiss-Prot fields, which include information
that in the future these, and other, alternative approaches
as varied as catalytic activity, quaternary structure, signal
will be important complements for research in protein
sequences, catalytic residues, domain structure, and post-
functions. However, homology-based function prediction
still plays a central role in Molecular Biology.
The current efforts for the construction of classifications
(ontologies) for the definition of function at various levels
The Space of Sequences and the Annotation of
(particularly the GO Consortium27) represent a possible
Function
way of alleviating these problems. The use of the current
Our system works by first mapping the sequence space
GO ontology in our (and other) automatic annotation
in groups of orthologous sequences. The results of the
methods will not be easy until a substantial number of
clustering depend strongly on the quality of the sequence
sequences from various genomes are annotated, some-
space map built. This, in turn, depends on the parameters
thing that had still not happened at the time these systems
used for retrieving the sequences and measuring the
were built, despite the considerable effort made by the
distances between them. We have shown previously23 that
this clustering strategy works appropriately for the identi-
Selecting Representative Descriptions
fication of orthologous groups of sequences from sets ofparalogues (families and subfamilies). Compared to other
Our purpose was to identify the most representative
approaches for protein classification, this one has the
description from a set of functionally related ones. We
advantage of being resistant to domain problems because
tested various ways of measuring homogeneity of the
sequence searches are made with respect to a query
descriptions. Both Shannon entropy and the sum of log-
protein and only the aligned subsequences are used to
probabilities (probabilities understood as the frequency of
search the space of sequences. Additionally, it does not
words) tend to give better scores to long descriptions, even
require working in the context of complete genomes, as in
if they contain poorly represented words. The comparison
of the frequency of words in a given description with the
The procedure has been validated in real biological
frequency in the whole set of descriptions using Relative
problems, such as the annotation of the Buchnera aphidi-
Entropy did not render useful descriptions. In our hands,
cola25 genome described here.
the best results were obtained by calculating the informa-
Systems able to make solid predictions of function based
tion content of the description weighted by the number of
on sequence information are important in the context of
words in the description. We additionally apply a correc-
the annotation of genomes, even if a number of difficulties
tion to avoid penalizing longer descriptions that include
remain to be solved. The definition of function has a very
important subjective component: the same protein will bedescribed in different terms by different scientists. The
Prediction of Function from Homologous
approach we have followed tries to transfer the most
Sequences and Alternative Approaches
representative description, the consensus definition, which
Interestingly, the annotation of function by transference
also reduces the pernicious propagation of annotation
from proteins of related sequences is not the only possibil-
errors. Domain shuffling events observable at many pro-
ity for the “in silico” prediction of function. The flourishing
teins make function transference based on homology dan-
of genomic data has enabled other modes of function
gerous. An analysis of domain structure is important but,
prediction independent of the identification of homologous
as properly expressed by Attwood,32 the complexity of
sequences. The function of proteins can be inferred from
biological systems (such as complete proteins) poses impor-
the study of the similarity of their expression pattern with
tant problems for computational approaches because the
properties of a system can be explained by but not deduced
Sander C. Automated genome sequence analysis and annotation.
from its components (such as protein domains). In this
14. Moller S, Leser U, Fleischmann W, Apweiler R. EDITtoTrEMBL:
work, we did not analyze the individual components but
a distributed approach to high-quality automated protein se-
rather, as a partial solution, assessed the extent to which
quence annotation. Bioinformatics 1999;15:219 –227.
the similarity covers the entire length of the implied
15. Fleischmann W, Moller S, Gateau A, Apweiler R. A novel method
for automatic functional annotation of proteins. Bioinformatics
16. Sigrist CJ, Cerutti L, Hulo N, Gattiker A, Falquet L, Pagni M,
ACKNOWLEDGMENTS
Bairoch A, Bucher P. PROSITE: a documented database using
We acknowledge the suggestions of O. Olmea for the
patterns and profiles as motif descriptors. Brief Bioinform 2002;3:265–274.
application of the clustering strategies, and the graph-
17. Bairoch A, Apweiler R. The SWISS-PROT protein sequence
based representation of the recursive search results. We
database and its supplement TrEMBL in 2000. Nucleic Acids Res
are grateful to members of the Protein Design Group for
18. Reich J, Mitchell A, Goble C, Attwood T. Toward more intelligent
interesting discussions and continuous support. Our work
annotation tools: a prototype. IEEE Intell Syst 2001;16:42–51.
has benefited from the interesting ideas on the Ncut
19. Li W, Jaroszewski L, Godzik A. Clustering of highly homologous
algorithm as described in G. Yona’s PhD work,33 and from
sequences to reduce the size of large protein database. Bioinformat-ics 2001;17:282–283.
use of the MESCHACH numerical library, made public by
20. Park J, Teichmann S, Hubbard T, Chothia C. Intermediate
D. E. Stewart and Z. Leyk. We thank Ian Korf for the
sequences increase the detection of homology between sequences.
BPlite BLAST parser. This work has in part been sup-
ported by a grant from the Spanish Ministry of Science and
21. Gerstein M. Measurement of the effectiveness of transitive se-
quence comparison, through a third ’intermediate’ sequence.
Technology (CICYT), and by a fellowship from Madrid’s
22. Shi J, Malik J. Normalized cuts and image segmentation. Proc
IEEE Conf Comp Vision Pattern Recog 1997;731–737. REFERENCES
23. Abascal F, Valencia A. Clustering of proximal sequence space for
the identification of protein families. Bioinformatics 2002;18:908 –
1. Altschul SF, Gish W. Local alignment statistics. Methods Enzymol
24. Wu Z, Leahy R. An optimal graph theoretic approach to data
2. Smith TF, Zhang X. The challenges of genome sequence annota-
clustering: theory and its application to image segmentation.
tion or “The devil is in the details”. Nature Biotechnol 1997;15:
25. van Ham RCHJ, Kamerbeek J, Palacios C, Rausell C, Abascal F,
3. Bork P, Dandekar T, Diaz-Lazcoz Y, Eisenhaber F, Huynen M,
Bastolla U, Ferna´ndez JM, Jime´nez L, Postigo M, Silva FJ,
Yuan Y. Predicting function: from genes to genomes and back. J
Tamames J, Viguera E, Latorre A, Valencia A, Mora´n F, Moya A.
Reductive genome evolution in buchnera aphidicola. Proc Natl
4. Bork P, Koonin EV. Predicting functions from protein sequences:
where are the bottlenecks?. Nature Genet 1998;18:313–318.
26. Tamas I, Klasson L, Canback B, Naslund AK, Eriksson AS,
5. Doerks T, Bairoch A, Bork P. Protein annotation: detective work
Wernegreen JJ, Sandstrom JP, Moran NA, Andersson SG. 50
for function prediction. Trends Genet 1998;14:248 –250.
million years of genomic stasis in endosymbiotic bacteria. Science
6. Andrade MA. Position-specific annotation of protein function
based on multiple homologs. ISMB 99 1999;7:28 –33.
27. The Gene Ontology Consortium. Gene ontology: tool for the
7. Todd AE, Orengo CA, Thornton JM. Evolution of function in
unification of biology. Nature Genet 2000;25:25–29.
protein superfamilies, from a structural perspective. J Mol Biol
28. Valencia A, Pazos F. Computational methods for the prediction of
protein interactions. Curr Opin Struct Biol 2002;12:368 –373.
8. Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller
29. Tatusov RL, Koonin EV, Lipman DJ. A genomic perspective on
W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation
protein families. Science 1997;278:631– 636.
of protein database search programs. Nucleic Acids Res 1997;25:
30. Marcotte EM, Pellegrini M, Thompson MJ, Yeates TO, Eisenberg
D. A combined algorithm for genome-wide prediction of protein
9. Brenner SE. Errors in genome annotation. Trends Genet 1999;15:
31. Jensen LJ, Gupta R, Blom N, Devos D, Tamames J, Kesmir C,
10. Devos D, Valencia A. Intrinsic errors in genome annotation.
Nielsen H, Staerfeldt HH, Rapacki K, Workman C, Andersen CA,
Knudsen S, Krogh A, Valencia A, Brunak S. Prediction of human
11. Devos D, Valencia A. Practical limits of function prediction.
protein function from post-translational modifications and localiza-
tion features. J Mol Biol 2002;319:1257–1265.
12. Blaschke C, Hirschman L, Valencia A. Information extraction in
32. Attwood TK. Genomics. The Babel of bioinformatics. Science
molecular biology. Brief Bioinform 2002;3:154 –165.
13. Andrade MA, Brown NP, Leroy C, Hoersch S, Daruvar A.De,
33. Yona G. Methods for global organization of the protein sequence
Reich C, Franchini A, Tamames J, Valencia A, Ouzounis C,
space. PhD Thesis, Hebrew University. 1999.
B.D.K.C SUPPLEMENTARY REGULATIONS CLUB CHAMPIONSHIPS 2011 Held at Ellough Park Raceway, Beccles, Suffolk, NR34 7XD During 2011 Beccles & District Kart Club Ltd (BDKC, the club) will organize National Clubman Permit meetings on the March, April, May, June, July, August, September, October and November. The meetings will be held under the general regulations of the Motor Sports Assoc
Hepatite aguda por Dengue CASE REPORT ACUTE HEPATITIS DUE TO DENGUE VIRUS IN A CHRONIC HEPATITIS PATIENT Souza L.J1, Coelho J.M.C.O.4, Silva E.J. 2, 5, Abukater M.1, 2, Almeida F.C.R.1, 2, Fonte A. S.1, 2, Souza L.A. 1,3 1Centro de Referência da Dengue/Hospital Plantadores de Cana – Campos dos Goytacazes – RJ; 2Faculdade de Medicina de Campos; 3Universidade Estácio de Sá;
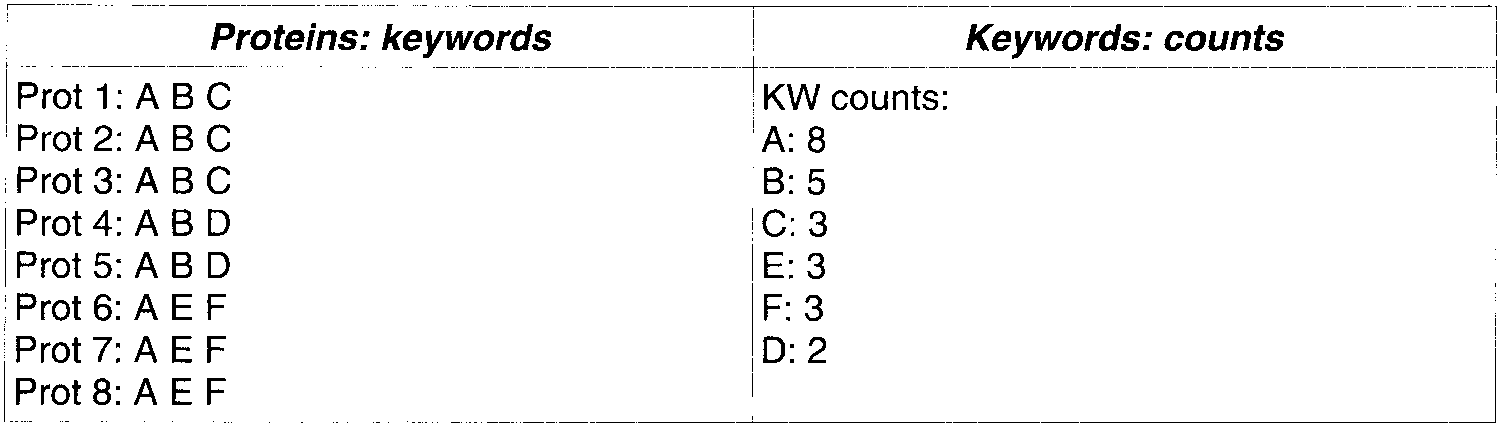 Key words accepted: A B C. Note that C, E, and F have the same frequency, but only C is transferred
to avoid mixing key words that do not co-occur. The process: first A is selected as seed. Then, since B isconnected to A more than 4 times (8/2), B will also be accepted. Then C will also be accepted because it isconnected to B more than 2.5 times (5/2). No more keywords will be added.
Key words accepted: A B C. Note that C, E, and F have the same frequency, but only C is transferred
to avoid mixing key words that do not co-occur. The process: first A is selected as seed. Then, since B isconnected to A more than 4 times (8/2), B will also be accepted. Then C will also be accepted because it isconnected to B more than 2.5 times (5/2). No more keywords will be added. (TET(S)) instead of TETM. This is an especially problem-atic case where the clustering is not able to classify intoseparate groups the different tetracycline resistance deter-minants. Instead, it puts them together according to theirhigh similarity. The annotations in the database (or thenomenclature) seem to be inconsistent (or the specificityhas no evolutionary foundation), because the percentage ofsequence identity is much higher between some Tet(M) andTet(whatever) than between two Tet(M), for example, TETM-_NEIME vs. TETS_LACLA (77%) and TETM_NEIME vs.
(TET(S)) instead of TETM. This is an especially problem-atic case where the clustering is not able to classify intoseparate groups the different tetracycline resistance deter-minants. Instead, it puts them together according to theirhigh similarity. The annotations in the database (or thenomenclature) seem to be inconsistent (or the specificityhas no evolutionary foundation), because the percentage ofsequence identity is much higher between some Tet(M) andTet(whatever) than between two Tet(M), for example, TETM-_NEIME vs. TETS_LACLA (77%) and TETM_NEIME vs.