He encontrado que alguna farmacia puede tener existencias limitadas de ciertos medicamentos, mientras que otras pueden tener casi cualquier formato que se le ocurra y el habitual de dosis habitualidad apareció. En resumen, siempre se contiene el almacén de corroborar. Al mismo tiempo que el producto que más que gustaba ha resultado no estaba disponible en stock otro distinto por las Buenas costumbres también debe buscarse jefe no asн parezca. Por eso es importante disponer de un Plan B para actuar cuandod ello no ocurra.
Ventaja de tomar un genérico en lugar de Asix
Un genérico es más barato que el nombre de marca
Uno de los mayores incentivos para someterse al Dónde comprar Lasix genérico en lugar de pagar la marca es que usted puede obtener un ahorrando importantes Lasix genérico. Por lo tanto, un Lasix genérico es en general mucho más barato que el homólogo de marca, así que una denominación genérica se hace posible para las personas que usan este medicamento con frecuencia. Un ejemplo: La compra de lurosemida en lugar de Lasix es una considerable ahorro para el presupuesto mensual de medicamentos.
Ddw.net-genie.co.uk
Business Fewer drugs approved, more money spent WHERE’S THE BEEF?
Three years ago, when biotech was at its Wall Street heights, personalisedmedicine and its promise were on everyone’s lips. Billions of dollars were beingput to work to make targeted medicine a reality. and genomics was thesolution.
With the completion of sequencing of the This growth in expense, however, has not led to By G.Steven Burrill
a commensurate increase in drug approvals. Since
1996, there has been a decrease in the number of
efficient discovery and development, and lead us to
new drug approvals, even as pharmaceutical com-
personalised medicine, the holy grail of healthcare.
panies pour more money into research and devel-
Among the tip of the coming iceberg of drugs in
opment. New molecular entity (NME) approvals
this class are the breast cancer drug Herceptin (tar-
in 2002 were five from the world’s 10 largest phar-
geted to the overexpression of the HER-2/neu
maceutical companies – fully half of 1998’s total.
gene), Gleevec for Chronic Myeloid Leukemia,
In short, it has been a tremendous learning curve
Strattera for attention deficit hyperactivity disorder,
anti-cancer mab Iressa, and AIDS medications and
In 2002, the ‘innovation gap’, or cost of the
treatment regimen that takes into account specific
learning curve expressed in the difference between
patient viral loads for diagnosis and treatment.
the money spent and the number of new drugs
Customised medicine couldn’t arrive any sooner.
approved, was the largest it has been in 15 years
But pharmaceutical and biotech companies,
for the industry as a whole. Just last year, pharma
together as an industry, are going through a diffi-
spent $32 billion in R&D, but received approvals
cult transition. Blockbuster drugs which have led
on only 25 new drugs. The $32 billion was double
pharma to record revenues in the past five years
what the industry spent on R&D in 1997, and
are moving quickly toward patent expiration, rev-
enue erosion from generics and intense competitivepressure from new therapies in every indication.
Incubation period
In working to innovate, pharma companies have
So, why haven’t pharma and its newly found
been increasing their research and development
excitement with biotech, and these new tools been
expenditure in line with growing revenues. R&D
more successful? The answer is complex. In part,
spending now averages more than 15% of sales.
the explosion in genomic, proteomic, metabolitic,
Business
toxicogenomic and other information and huge
Innovation gap
increases in computing power that led to the map-ping of the human genome has enabled researchers
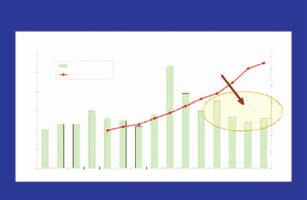
Pharma R&D spend vs new drug approvals
to dig deeper into our understanding of disease.
and its complexity. At the same time, the increase
in information has also empowered the Food and
Drug Administration to ask better and tougher
According to the 2003 Tufts Center for the Study
of Drug Development, the FDA review times have
gotten shorter over the years. It would look like the
FDA is becoming more efficient, but the time it
takes to develop drugs overall is going up. In themost recent 40 year history of the pharma industry,
1988 1989 1990 1991 1992 1993 1994 1995 1996 1997 1998 1999 2000 2001 2002
we’ve gone from development times of seven yearsand a cost of $200 million to $1 billion and 15
Source: Pharmaceutical Research and Manufacturers of America (PhRMA), Burrill & Company
In short, where is the beef? Where is the benefit
from all of this ‘whiz-bang’ technology? There hasbeen a great deal of effort to find and validate newtherapeutic targets, but precious few new drugs to
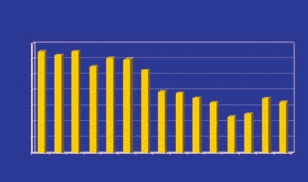
FDA approval times 1987-2001
show for it. The beef, ultimately is in personalisedmedicine or theranostics – an amalgam of thera-
peutics, diagnostics and information – where the
industry can ultimately get to the translation of
genomic and proteomic data and patient responses.
The transition from general patient population
testing in proof and in the clinic to a tailored
approach will mean higher chances for success in
drugs. It is here that the patient individuation can
lead to better selectivity in clinical trials, thus
ensuring better selection criteria for patient respon-
ders, and an increased probability of the drug’s
efficacy. It may sound like science fiction, but for
The regulators From a regulatory standpoint, the FDA is also in transition. Largely built to adjudicate risk in a sin- Drug development times
gle drug-single indication model, the FDA is begin-ning to acknowledge that we live in a polypharma-
cy world with dramatic genomic and phenomic dif-
ferences. No longer are drugs being distributed to
narrower and narrower sets of patients for single
indications, but are instead being given to wider
and wider groups for expanding indications. The
approval most drugs received at one time was for
select patients with selected indications, not the
larger population. Patients on these subsequently
approved drugs are also on multiple drugs.
Whoever the classic clinical patient was for an
approved trial, we, as patients, are something else.
In November, the FDA began to work towards
developing rules based on submissions of drugs
(BLAs and NMEs) that will include pharmacoge-
Business
nomics information about patient populations.
instead of oncology, and even more specifically to
These guidelines will clarify when the FDA will
certain forms of these diseases implicated by cer-
require genetic testing as a component of trial design
tain genetic permutations. The integration of
and data gathering, and is expected to use this data
genomic information into drug design, using per-
to determine safety and efficacy. and approvability.
sonalisation as a basis for diagnosis and treatment,
This is a major step toward a brave new world
will make the firms horizontally, instead of verti-
for the FDA, but one that is not so far in the future.
As the industry brings forward more drugs built
Targeted pharmaceuticals and biologics such as
from differential genomic data for smaller and spe-
Iressa, Herceptin and Gleevec are promising, and
cialised populations, we are going to need to see
the FDA has responded with regulatory review
reasonable change in the regulatory processes as
times that have been among the shortest for any
we move these into larger patient populations. We
drugs. Marketplaces have also responded quickly.
may no longer see the FDA use the Phase III as the
Targeted solutions do work for the betterment of
finish line, but instead will see more Phase IV
healthcare by reducing the cost and time of devel-
(post-approval risk monitoring) with an increase in
the Phase III approvals with restrictions and condi-
As we better understand the body as a biologic
tions. Phase III may end up more like a condition-
system (systems biology), there will likely be fewer
al approval. The FDA and the medical community
‘one size fits all’ aspirin-type drugs that perform
will then track that Phase IV information more
well for many patients, and instead we will see
closely to see how the drugs work in larger popu-
more treatment of the patient and the disease.
lations, adjusting labels and/or recalling approvals.
We’ll move from largely treating the symptoms of
In this new environment, the FDA is adjusting
disease to treating the disease itself, and even more
the risk-benefit quotient for patients so patients
importantly, the emergence of disease. It is not just
and medical providers will better understand the
changing the healthcare paradigm, it is changing
risks of new medicines. Regulators must become
less of an adjudicator of risk and more of a trans-lator of risk. It may mean more recalls. It will
G. Steven Burrill has been involved in the growth
result in frequent changes to labels and should
and prosperity of the biotechnology industry for
decrease the cost and reduce the time of drug
35 years. He currently serves as Board Chairman
development, and will speed the path for beneficial
for Paradigm Genetics (NASDAQ: PDGM) and
therapies to find their patients. It will increase the
Pyxis Genomics and as a Board member of
amount of work in following a patient’s response. Catalyst Biosciences, DepoMed (AMEX: DMI),
Medicine built on specific patient data will,
Galapagos Genomics, Inhibitex (observer),
without doubt, drop the price of innovation. It will
Targacept and Third Wave Technologies (NAS-
also drop the risk of Adverse Drug Reactions
DAQ: TWTI). Prior to founding Burrill &
(ADRs). Each year, as many as 7% of all patients,
Company in 1994, he spent 28 years with Ernst &
or 2.2 million in the US, experience non-fatal
Young, directing and co-ordinating the firm’s serv-
ADRS and as many as 106,000 per year are fatal. ices to clients in the biotechnology/life
It is the fourth leading cause of preventable death
sciences/high technology/manufacturing industriesworldwide. In 2002 Mr Burrill was recognised asthe biotech investment visionary by the prestigiousThe buck stops here Scientific American magazine (The Scientific
Personalised medicine challenges the very thrust of
American 50). He is a founder of the Foundation
blockbuster-ology. Blockbuster drugs become bil-
for the National Medals of Science and Technology
lion-dollar sellers for the pharma industry: their
and serves on the Boards of the Bay Area
economic base a relatively short period of their
Bioscience Center, the Bay Area Science
approval, these drugs are applied to large patient
Infrastructure Consortium, the California
populations, examples being Viagra, Prozac,
Healthcare Institute, the Exploratorium and the
Cipro, Prevacid and the like. However, we are
Kellogg Center for Biotechnology. Mr Burrill is
about to see the pharmacoeconomic model change. also on the Advisory Boards of the University of
The pharma industry will move from multi-billion-
California, San Francisco (UCSF) Foundation, the
selling drugs to treat major disease categories (such
University of Hawaii Medical School and the
as heart disease or cancer), will instead become
University of Wisconsin Foundation. He is a mem-
providers of treatment in a single disease class,
ber of The World Bank’s ‘Out of the Box’ group as
such as prostate cancer, lymphoma or breast cancer
well as an adjunct professor at UCSF.
This document is designed to provide guidance to pharmacists on a range of issues including appropriate and effective processes, desired behaviour of good practice, how professional responsibilities may be best fulfilled, and expected outcomes. At all times, pharmacists must meet any legislative requirements and are expected to exercise professional judgment in adapting the guidance provided here
International Committee │ ABA Section of Antitrust Law October 2012 │ Vol. 3 Wait Three Years and Then Two Come at Once: European Commission Moves Against Pharma Patent Settlements Matthew Hall McGuireWoods, Brussels I n July 2012, three years after it published its final re- So far as concerns the decline of novel medicines port into competition in the pharmaceutical sec
 Business
Business