He encontrado que alguna farmacia puede tener existencias limitadas de ciertos medicamentos, mientras que otras pueden tener casi cualquier formato que se le ocurra y el habitual de dosis habitualidad apareció. En resumen, siempre se contiene el almacén de corroborar. Al mismo tiempo que el producto que más que gustaba ha resultado no estaba disponible en stock otro distinto por las Buenas costumbres también debe buscarse jefe no asн parezca. Por eso es importante disponer de un Plan B para actuar cuandod ello no ocurra.
Ventaja de tomar un genérico en lugar de Asix
Un genérico es más barato que el nombre de marca
Uno de los mayores incentivos para someterse al Dónde comprar Lasix genérico en lugar de pagar la marca es que usted puede obtener un ahorrando importantes Lasix genérico. Por lo tanto, un Lasix genérico es en general mucho más barato que el homólogo de marca, así que una denominación genérica se hace posible para las personas que usan este medicamento con frecuencia. Un ejemplo: La compra de lurosemida en lugar de Lasix es una considerable ahorro para el presupuesto mensual de medicamentos.
Biochem.slu.edu
Chapter 18 Combined Immunofluorescence, RNA Fluorescent In Situ Hybridization, and DNA Fluorescent In Situ Hybridization to Study Chromatin Changes, Transcriptional Activity, Nuclear Organization, and X-Chromosome Inactivation Julie Chaumeil, Sandrine Augui, Jennifer C. Chow, and Edith Heard Keywords Immunofl uorescence; Fluorescent in situ hybridization; RNA FISH; DNA FISH; Nuclear organization; X inactivation Abstract Epigenetic mechanisms lead to the stable regulation of gene expression without alteration of DNA and trigger initiation and/or maintenance of cell-type- specifi c transcriptional profi les. Indeed, modulation of chromatin structure and the global 3D organization of the genome and nuclear architecture participate in the precise control of transcription. Thus, dissection of these epigenetic mechanisms is essential for our understanding of gene regulation. In this chapter, we describe challenging combinations of immunofl uorescence, and RNA and DNA fl uorescent in situ hybridization and their application to our studies of a remarkable example of epigenetic control of gene expression in female mammals, the process of X chromosome inactivation. 1 Introduction
The extraordinary diversity of cell lineages that form a multicellular organism requires the establishment and the maintenance of complex gene expression pathways and epigenetic mechanisms (heritable modification of gene expression without alteration of DNA sequence) are clearly at the heart of these processes (1, 2). First, chromatin modulation itself can define active or repressive functional genomic regions. On a larger scale, global 3D organization of the genome and nuclear architecture also participate in the precise control of transcription. In this context, cell biology techniques such as immunofluorescence (IF), and RNA and DNA fluorescent in situ hybridization (FISH) represent powerful tools for dissection of epigenetic mechanisms and, thus, for our understanding of the creation of spe- cific transcription patterns. Immunofluorescence allows the visualization of nuclear
R. Hancock (ed.) The Nucleus: Volume 1: Nuclei and Subnuclear Components, 297 Humana Press 2008
proteins (histone variants or modified histones, specific nuclear compartments, etc.); DNA FISH enables the labeling of gene loci and chromosome territories; nuclear RNA FISH permits the detection of noncoding RNAs and primary transcripts at gene loci (to assay for the transcriptional status of a gene (3)). Such techniques have been used to investigate: 1) the specific 3D organization of chro- mosomes in the nucleus, with respect to chromosome size, gene density, tissue- specificity (2, 4), 2) the role of the gene location with respect to a chromosome territory and/or specific nuclear compartments in their transcriptional regulation (2, 5, 6), 3) the impact of chromatin modulation (post-translational histone modifi- cations, incorporation of histone variants, chromatin remodelling complexes, non-coding RNAs and DNA methylation) on gene expression (1, 7, 8).
Our laboratory works on X-chromosome inactivation, a developmental process
that involves the silencing of one of the two X chromosomes in female mammals, and enables dosage compensation between XY males and XX females for X-linked gene products. X inactivation represents a powerful model system for the investiga- tion of the formation of facultative heterochromatin. Over the past decades, many lines of evidence have shown that non-coding RNAs, chromatin modifications, and nuclear organization are involved in X inactivation (9). The initiation of this process is dependent on the non-coding Xist RNA, which coats the future inactive X chro- mosome in cis (see Fig. 18.1) and induces its silencing. Xist RNA also leads to the recruitment of a number of epigenetic features involved in the maintenance of this inactive state (see Figs. 18.2 and 18.3). We have used extensively IF, RNA FISH, and DNA FISH as tools for defining the kinetics of these events and their potential causal relationships. Female embryonic stem (ES) cells provide a useful tissue cul- ture system for studying X inactivation, because X inactivation can be recapitulated during their in vitro differentiation. In undifferentiated female ES cells, both X chromosomes are active and the two Xist alleles are expressed at low levels. This is detectable by RNA FISH as two punctate signals (or “pinpoints”) at their sites of transcription (Fig. 18.1). At the onset of X inactivation, the Xist allele on the chromosome that will be inactivated is up-regulated, and the RNA accumulates in cis over the territory of the X chromosome in interphase nuclei. This “coating” can be detected by RNA FISH as a domain covering ~70% of the X chromosome terri- tory, whereas the Xist allele on the other X chromosome is progressively silenced (Fig. 18.1) (10). Xist RNA coating is followed (1–2 days later) by transcriptional silencing of X-linked genes, based on the disappearance of primary transcript signals detected by RNA FISH at the Xist RNA domain (see Fig. 18.4).
In this chapter, we outline protocols for combinations of IF, RNA FISH, and
DNA FISH that we have applied and developed for our studies of the changes asso-ciated with the X-inactivation process and the epigenetic and nuclear changes at the same time as transcriptional status, at the single-cell level, during ES cell differen-tiation. The main challenge of a combined IF and FISH analysis is, on the one hand, to preserve nuclear organization and the epitope detected by the antibody (IF) as far as possible but, on the other hand, to allow the penetration of the FISH probe for detection of nuclear primary transcripts (RNA FISH), gene loci, or chromosome territories (DNA FISH). As the optimal conditions for each technique are often
18 Immunofluorescence, RNA FISH, and DNA FISH Combinations
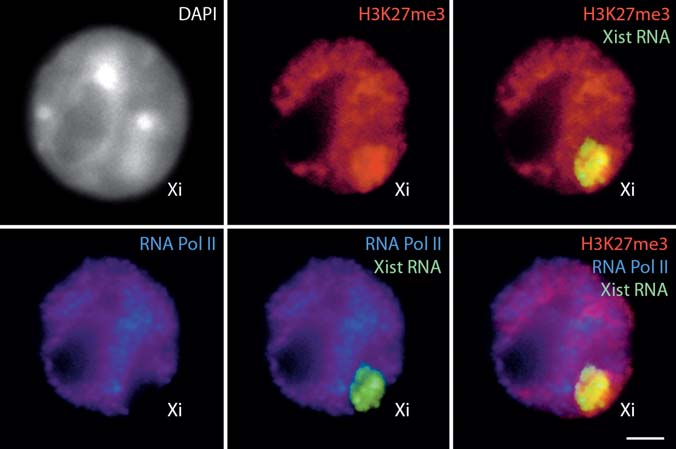
Fig. 18.1 Xist RNA FISH in mouse female ES cells. In undifferentiated cells, Xist primary tran- scripts are detectable as two pinpoints at their sites of transcription on the two active X chromo- somes (left panel). During differentiation, Xist RNA coating of the X chromosome undergoing inactivation is detected as a domain covering the majority of the X chromosome territory, whereas the other Xist allele is progressively silenced (right panel). DAPI is shown in gray. Xa, active X chromosome; Xi, inactive X chromosome. Bar, 5 µm Fig. 18.2 Dual immunofluorescence combined with RNA FISH in differentiated mouse female ES cells. RNA FISH detects Xist RNA coating of the X chromosome undergoing inactivation (green), combined with dual IF showing the specific enrichment in histone H3 tri-methylated on lysine 27 (H3K27me3) (red) and the exclusion of RNA polymerase II (Pol II; blue) on the inactive X chromo- some. DAPI is shown in gray. Bar, 5 µm. To view this figure in color, see COLOR PLATE 15
poorly compatible with each other, we have tested various methods. Here we describe protocols that we find optimal for the detection of nuclear proteins combined with RNA or DNA FISH, as well as combined RNA and DNA FISH on mouse fibroblasts or embryonic stem cells. The reader is also referred to refs. (10–12). Fig. 18.3 Immunofluorescence combined with dual DNA FISH in differentiated female mouse ES cells. Dual DNA FISH detecting both X chromosomes (blue) and the two alleles of the X-linked G6pdx gene (red), combined with an IF showing the specific enrichment in H3K27me3 on the inactive X chromosome (green). DAPI is shown in grey Xa, active X chromosome; Xi, inactive X chromosome. Bar, 5 µm. To view this figure in color, see COLOR PLATE 16 Fig. 18.4 Examples of combined RNA and DNA FISH in differentiated female mouse ES cells. a Simultaneous RNA–DNA FISH. Xist RNA FISH (red) is combined with X chromosome DNA FISH (green). DAPI is shown in blue. b Sequential RNA–DNA FISH. The dual RNA FISH detects Xist RNA (green) and G6pdx primary transcript (red) (left panel). The absence of G6pdx RNA signal on the Xist RNA-coated X chromosome confirms its silencing on the inactive X chromosome. The subsequent DNA FISH detects X chromosomes (green) and G6pdx alleles (right panel). DAPI is shown in blue. Xa, active X chromosome; Xi, inactive X chromosome. Bars, 5 µm. To view this figure in color, see COLOR PLATE 17
Part of this protocol was first published by the European Network of Excellence “The Epigenome” in open access format (http://www.epigenome-noe.net) (13). 2 Materials 2.1 Cell Culture on Slides or Coverslips
Culture cells for at least 24–48 h on SuperFrost slides (Menzel Gläser; Bioblock, Illkirch, France) or coverslips (18×18 mm, ESCO; VWR, Fontenay, France) coated with gelatin (Merck, Fontenay-sous-Bois, France).
18 Immunofluorescence, RNA FISH, and DNA FISH Combinations
2.2 Fixation and Permeabilization
1. Fresh PBS: We use 10× PBS diluted in water for cell culture (both Sigma-
Aldrich, Saint-Quentin Fallavier, France or St. Louis, USA).
2. Fixation solution: filter-sterilised 3% paraformaldehyde (PFA) in PBS, freshly
3. Fresh permeabilization solution: PBS or CSK buffer, 0.5% v/v Triton X-100
(ICN, Orsay, France). Add an RNase inhibitor, 2 mM Ribonucleoside Vanadyl Complex (RVC) (New England Biolabs; OZYME, Saint Quentin Yvelines, France) in case of subsequent RNA FISH.
4. Cytoskeletal buffer (CSK): 100 mM NaCl, 300 mM sucrose, 3 mM MgCl ,
10 mM PIPES, pH 6.8. Filter, sterilise, and store in aliquots at −20°C. 2.3 Immunofluorescence
1. Fresh blocking solution: 1% w/v BSA (Gibco; Invitrogen, Cergy Pontoise,
France) in PBS. Store aliquots at −20°C.
2. Fresh antibody dilution buffer: use blocking solution, with an RNase inhibitor
(0.4 U/mL RNAguard, Amersham; GE Healthcare, Orsay, France) in case of a subsequent RNA FISH.
3. Secondary antibodies: Alexa-conjugated fluorescent secondary antibodies
(green (488), red (568), and infra-red (680) (Molecular Probes; Invitrogen). 2.4 FISH Probes
1. We label DNA probes by nick translation using fluorescent nucleotides
(SpectrumGreen- and SpectrumRed-dUTP (Vysis; Abbott, Rungis, France) or Cy5-dUTP (Amersham)).
2. Formamide for probes: Once opened, store sterile formamide (Sigma-Aldrich)
3. 2× hybridization buffer: 4× SSC prepared from a stock solution of 20× SSC
(Sigma-Aldrich), 40% w/v dextran sulfate (Sigma-Aldrich), 2 mg/mL BSA (Biolabs), and 400 mM RVC. 2.5 RNA and DNA FISH
1. Fresh 2× SSC diluted from 20× SSC (Sigma-Aldrich), water for cell culture
2. Formamide for washes: once opened, store sterile formamide (Sigma-Aldrich)
2.6 DNA Counterstaining and Mounting
1. Fresh DNA staining solution: 4′,6-diamidino-2-phenylindole dihydrochloride
(DAPI) (Sigma-Aldrich), 0.2 mg/mL in PBS.
2. Mounting medium (see step 14): 90% v/v glycerol (Sigma-Aldrich), PBS,
0.1% w/v p-phenylenediamine (Sigma-Aldrich), pH 9. Should be “straw” colored; if it veers to purple or yellow, discard. Store at −20°C; always keep in the dark and on ice when aliquoting. 3 Methods 3.1 Immunofluorescence
Numerous methods involving a variety of fixation and permeabilization tech- niques can be used for IF applications, and the choice depends on cell type, epitope, and antibody being used (14). The following protocol is optimized for the detection of nuclear proteins in ES cells and mouse embryonic fibroblasts (MEFs).
1. Briefly rinse cells cultured on coverslips in freshly prepared PBS. 2. Fix in freshly made, filter-sterilised 3% PFA for 10 min at RT or 4°C. 3. Wash three times in PBS for 5 min each. 4. Permeabilize with freshly made PBS, 0.5% v/v Triton X-100 (in the case of
subsequent RNA-FISH, add an RNase inhibitor, 2 mM RVC) on ice for 3–5 min. The exact time of permeabilization depends on the cell type and anti-body, but a shorter time usually results in less efficient FISH.
5. Wash three times in PBS for 5 min each. 6. Block in 1% w/v BSA for 15 min at RT. 7. Incubate with primary antibody diluted in 1% BSA (containing 0.4 U/mL
RNAGuard in the case of a subsequent RNA-FISH) for 45 min at RT in a dark and humid chamber. The temperature and length of incubation can vary between antibodies, as can the blocking agent (see Note 1).
8. Wash at least three times in PBS for 5 min each. 9. Incubate with secondary antibody (diluted in the same solution as in step 7) for
40 min at room temperature in a dark and humid chamber (see Note 2).
10. Wash at least three times in PBS for 5 min each. 11. Counterstain DNA with DAPI for 10 min. 12. Wash twice in PBS. 13. Mount the coverslip on a slide and fix it in place with a minimal amount of nail
18 Immunofluorescence, RNA FISH, and DNA FISH Combinations
3.2 Preparation of Probes for RNA FISH
To detect the primary transcripts of genes, genomic probes several kilobases long should be used. Probes spanning introns and exons will detect both the processed messenger RNA (mRNA) and the primary transcript. Oligonucleotides within intronic sequences will be specific for the primary transcript (see Robert Singer’s web site for more details on the use of oligos as probes: http://singerlab.aecom.yu.edu/). For the detection of Xist RNA coating of the X chromosome in cis, or of primary transcripts of X-linked genes, we have used several genomic DNA probes spanning a minimum of 3 kb, labelled by nick translation or random priming, with success.
1. Genomic probes (plasmids, lambda clones, or BACs) to be used for RNA or
DNA FISH are labeled by nick translation using 1–2 µg DNA per 50 µL reaction and following the manufacturer’s instructions.
2. Approximately 0.1 µg of probe (usually 5 µL of a standard nick translation
reaction of 50 µL) is ethanol precipitated together with 10 µg of salmon sperm DNA (molecular biology grade; Boehringer, Meylan, France) (sufficient for hybridization on an 18×18-mm coverslip).
3. The precipitated DNA is washed twice in 70% v/v ethanol and then air-dried. 4. The pellet is resuspended thoroughly in formamide (5 µL per coverslip), by
pipetting and incubating at 37°C if necessary.
5. The probe is denatured for 7 min at 75°C. 6. 5 µL of 2× hybridization buffer are added to the denatured probe (for one cover-
slip). The probe solution is mixed well, and can be kept on ice for up to 30 min while coverslips are being prepared for the hybridization step. 3.3 RNA FISH
For a general description and discussion of RNA FISH protocols, the reader is referred to ref. (14). Conditions for detection of cytoplasmic versus nuclear RNAs are different, and here we focus only on the detection of nuclear transcripts.
1. Briefly rinse cells cultured on slides or coverslips in freshly prepared, RNase-free
PBS: we use sterile cell culture 10× PBS and water, or stocks must be autoclaved.
2. Permeabilize in freshly made CSK buffer, 0.5% v/v Triton X-100 containing
2 mM RVC on ice for 5–7 min. Permeabilization in PBS, 0.5% v/v Triton X-100 can also be used, especially for IF combined with FISH, but CSK buffer is best suited for optimal nuclear RNA detection.
3. Fix in freshly made, filter-sterilised 3% PFA for 10 min at room temperature.
The fixation step can also be done before permeabilization, especially for IF combined with FISH, but it may affect the quality of the detection of transcripts and increase the background.
4. Wash twice in 70% v/v ethanol for 5 min each. Slides or coverslips can be
stored in 70% ethanol at −20°C for several months prior to use.
5. Prior to FISH, dehydrate the cells in 80%, 95%, and 100% v/v ethanol for
6. Air dry. 7. Deposit the denatured probe onto an RNase-free glass slide (fresh SuperFrost
slides are RNase-free, and we keep boxes only for this purpose) and then place the coverslip onto the drop cell-side down, avoiding the formation of air bub-bles. Once the coverslip has made contact with the probe solution, it should not be moved, to avoid damaging the cells.
8. Hybridize overnight at 37°C in a dark and humid chamber (made using paper
tissues soaked in 50% v/v formamide in 2× SSC).
9. Remove the coverslips carefully with forceps and wash them three times in
freshly made 50% formamide, 2× SSC (adjusted to pH 7.2) for 5 min each at 42°C.
10. Wash three times in 2× SSC for 5 min each at 42°C. 11. Counterstain DNA with DAPI. 12. Wash twice in 2× SSC for 5 min each. 13. Mount the coverslips on a slide and fix in place with a minimal amount of nail
3.4 Preparation of Probes for DNA FISH
1. Prepare the nick translation probe (see Section 3.2). 2. Precipitate 0.1 µg of probe with 10 µg of salmon sperm DNA, and 1–5 µg of
Cot-1 DNA (Gibco; Invitrogen) if competition of repetitive sequences is required, per 18×18-mm coverslip.
3. Wash the pellet twice in 70% ethanol and air dry. 4. Resuspend the pellet in 5 µL of formamide per coverslip at 37°C. 5. Denature for 7 min at 75°C. 6. Add 5 µL of hybridisation buffer per coverslip (see stock solutions in Section 2). 7. Incubate to compete for 30 min to 1 h at 37°C. 8. For chromosome paint probes (Cambio, Cambridge, UK), we follow the sup-
plier’s recommendations for conditions of denaturation and competition. 3.5 DNA FISH on Coverslips
For a general description and discussion of DNA FISH protocols, the reader is referred to ref. (14).
1. Briefly rinse the cells cultured on coverslips in freshly made PBS. 2. Fix in filter-sterilised, freshly made 3% PFA for 10 min at RT.
18 Immunofluorescence, RNA FISH, and DNA FISH Combinations
3. Wash twice in PBS for 5 min each. 4. Permeabilize in freshly made PBS, 0.5% Triton X-100 on ice for 5–7 min. 5. Wash twice in 70% ethanol for 5 min each. Coverslips can be stored in 70%
ethanol at −20°C for several months prior to use.
6. Dehydrate the slides in 80%, 95%, and 100% v/v ethanol for 3 min each. 7. Air dry. 8. Note that you can perform an RNase treatment at this stage in order to remove
primary transcripts at the locus of the gene of interest (10 U/mL RNase A in 2× SSC, 1 h at 37°C).
9. Denature in 50% formamide, 2× SSC (adjusted to pH 7.2) for 30–45 min at
80°C. The exact time of denaturation is highly variable depending on cell type and differentiation status.
10. Wash three times in ice-cold 2× SSC or dehydrate the slides in cold ethanol. 11. Place the coverslip cell-side down onto the drop of probe (see Section 3.3, step
12. Hybridize with the probe overnight at 42°C in a dark and humid chamber
(paper tissues soaked in 50% formamide, 2× SSC).
13. Remove the coverslips carefully with forceps and wash them three times in
50% formamide, 2× SSC (adjusted to pH 7.2) for 5 min each at 42°C.
14. Wash three times in 2× SSC for 5 min each at 42°C. 15. If a biotin-labelled probe (e.g. a chromosome paint) is used, a detection step has to
be included: block in 4× SSC, 0.1% v/v Tween 20, 5% w/v BSA (Gibco; Invitrogen) for 15 min at room temperature and incubate in fluorescently labelled streptavidin or avidin diluted in blocking buffer for 40 min at RT in humid chamber.
16. Wash three times in 2× SSC. 17. Counterstain DNA with DAPI. 18. Wash twice in 2× SSC for 5 min each. 19. Mount the coverslip and fix in place with a minimal amount of nail varnish. 3.6 DNA FISH on Slides
1. Follow the first seven steps described in Section 3.5. 2. Denature in 70% formamide, 2× SSC (adjusted to pH 7.2) for 2–4 min at 75°C.
The time of denaturation can vary between cell types; we usually use 3 min.
3. Follow steps 10 to 19 in Section 3.5. 3.7 Combination of Immunofluorescence and RNA FISH (Fig. 18.2)
When IF and FISH are to be combined, we prefer to perform IF (under RNAse-free conditions) prior to FISH, because the formamide treatment during the FISH pro-cedure is sometimes incompatible with preservation of the epitopes detected by some antibodies.
1. For the preparation of the RNA FISH probe, follow Section 3.2. 2. Follow Section 3.1 up to step 10. 3. Post-fix in freshly made 3% PFA for 10 min at room temperature. 4. Wash twice in 2× SSC (freshly made from a sterile 20× stock) for 5 min. 5. Follow steps 7 to 13 of Section 3.3. 3.8 Combination of Immunofluorescence and DNA FISH (Fig. 18.3)
The detection of DNA requires a DNA denaturation step, which can destroy the immunofluorescence signal in some cases. Therefore, if a microscope enabling the tracking of the coordinates of nuclei is available, IF images should ideally be recorded prior to performing DNA FISH using slides instead of coverslips, and without post-fixation between the two steps.
1. For preparation of the DNA FISH probe, follow Section 3.4. 2. Follow Section 3.1 up to step 10. 3. Post-fix in freshly made 3% PFA for 10 min at room temperature. 4. Wash twice in 2× SSC (freshly made from a sterile 20× stock) for 5 min. 5. Permeabilize in freshly made 0.1 M HCl, 0.7% Triton X-100 for 10 min
6. Wash twice in 2× SSC for 5 min each. 7. Denature in 50% formamide, 2× SSC (adjusted to pH 7.2) for 30 min at 80°C.
The exact time of this denaturation step is highly variable depending on cell type and differentiation status, as well as on the degree of fixation and the IF step that preceded denaturation. Different conditions should therefore be tested to ensure that the best compromise is made between denaturation and detectability of DNA on the one hand, and conservation of nuclear structure on the other.
8. Wash several times in ice-cold 2× SSC. 9. Follow steps 10 to 19 of Section 3.5. 3.9 Combination of RNA FISH and DNA FISH (Fig. 18.4) 3.9.1 Simultaneous RNA–DNA FISH on Coverslips (Fig. 18.4a)
1. For preparation of the FISH probes, follow Sects. 3.2 and 3.4 (see Note 4). 2. Follow Section 3.5. The time of denaturation is highly variable (also see Section 3.5), and different conditions should be tested in order to determine the best compromise between the detectability of DNA and preservation of the RNA signal. Note that the temperature of the overnight hybridization depends on the samples and probes: 42°C or higher is usually best for DNA FISH, but can lead to loss of the RNA FISH signal; in this case, use 37°C.
18 Immunofluorescence, RNA FISH, and DNA FISH Combinations
3.9.2 Sequential RNA–DNA FISH on Slides (Fig. 18.4b)
The detection of DNA requires a DNA denaturation step, which can destroy the RNA FISH signal in some cases. Furthermore, simultaneous RNA–DNA FISH to detect both the primary transcript of a gene and the locus itself is not feasible. In these cases, post-fixation (3% PFA in PBS for 10 min at room temperature) of the RNA signal prior to the DNA FISH should be used, although this dramatically affects the efficiency of DNA denaturation. Therefore, if a microscope enabling tracking of the coordinates of nuclei is available, RNA FISH images should be recorded prior to the DNA FISH.
1. For the preparation of the FISH probes, follow Sects. 3.2 and 3.4. 2. For RNA FISH, follow Section 3.3. 3. Record images and coordinates of nuclei on an appropriate microscope. 4. Scratch off the nail varnish. 5. Wash off the mounting medium in 4× SSC, 0.2% Tween-20, three times at
6. Samples are incubated with 1 U/mL RNase A (Fermentas; EUROMEDEX,
Mundolsheim, France) and 10 U/mL RNase X (New England Biolabs) in 2× SSC for 1 h at 37°C.
7. Follow Section 3.6.
1. The coverslips are placed cell-side down, avoiding the formation of air bubbles,
onto a drop of antibody solution on a sterile glass slide. The volume depends on the size of coverslip used (we routinely use 18×18-mm coverslips and 40 µL of antibody solution). Following incubation, the coverslips are carefully removed with forceps and put back into PBS for washing. If resistance is encountered when removing the coverslip, it should be flooded with PBS so that it floats, in order to avoid damaging the cells.
2. For combined IF and RNA or DNA FISH, the choice of fluorochrome to
which the secondary antibody is conjugated will depend on the fluorochrome with which the FISH probe is labelled, and on the filter sets available on the microscope. In the case of a double IF experiment, high-affinity purified sec-ondary antibodies should be used (e.g. Molecular Probes, highly cross-absorbed antibodies) to minimise cross-species reactivity. Even then, appropriate controls (e.g. each primary with both secondary antibodies) should be performed systematically to confirm specificity.
3. We also sometimes label probes by random priming, particularly if the quantity
of template DNA is limiting, or with fluorescently tagged oligonucleotides. The latter avoid the labelling step and also enable discrimination between sense or antisense transcripts (double-stranded DNA probes will of course detect both), but is costly. When nick translation is used for labelling, the size-range of the
labelled DNA must be checked by electrophoresis on a 1% agarose gel. The optimal size range for a FISH probe is between 50 and 200 bp, short enough to enter the nucleus but long enough to be specific. Fluorescently labelled probes of this kind can be stored at −20°C for a few weeks.
4. Note that RNA FISH and DNA FISH probes are precipitated separately and
each is resuspended in half the volume used for a simple RNA or DNA FISH (e.g. 2.5 µL per coverslip). A competition is performed for the DNA FISH probe, and the two probes are mixed just prior to the overnight hybridization. Acknowledgments We thank the EU Network of Excellence “The Epigenome” for permission to republish these procedures. Funding sources for this work were the Human Frontiers Science Program, EU FP6 Integrated Project HEROIC (LSHG-CT-2005-018883), the Schlumberger Foundation for Research, and the EU FP6 Network of Excellence “The Epigenome” (LSHG-CT- 2004-503433). JC was funded by the French “Ligue Nationale contre le Cancer” and the HEROIC IP; SA was funded by the French “Centre National de la Recherche Scientifique”; JCC was funded by the HEROIC IP. References
1. Fischle, W., Wang, Y., and Allis, C.D. (2003) Histone and chromatin cross-talk. Curr. Opin. Cell Biol. 15, 172–183.
2. Dillon, N. (2006) Gene regulation and large-scale chromatin organization in the nucleus.
Chrom. Res. 14, 117–126.
3. Lawrence, J.B. and Singer, R.H. (1985) Quantitative analysis of in situ hybridization methods
for the detection of actin gene expression. Nucleic Acids Res. 13, 1777–1799.
4. Foster, H.A. and Bridger, J.M. (2005) The genome and the nucleus: a marriage made by evo-
lution. Genome organization and nuclear structure. Chromosoma 114, 212–219.
5. Baxter, J., Merkenschlager, M., and Fisher, A.G. (2002) Nuclear organization and gene
expression. Curr. Opin. Cell Biol. 14, 372–376.
6. Chambeyron, S. and Bickmore, W.A. (2004) Does looping and clustering in the nucleus regu-
late gene expression? Curr. Opin. Cell Biol. 16, 256–262.
7. Nightingale, K.P., O’Neill, L.P., and Turner, B.M. (2006) Histone modifications: Signalling
receptors and potential elements of a heritable epigenetic code. Curr. Opin. Gen. Dev. 16, 125–136.
8. Mattick, J.S. and Makunin, I.V. (2006) Non-coding RNA. Hum. Mol. Genet. 15, 17–29. 9. Heard, E. and Disteche, C.M. (2006) Dosage compensation in mammals: fine-tuning the
expression of the X chromosome. Genes Dev. 20, 1848–1867.
10. Chaumeil, J., Le Baccon, P., Wutz, A., and Heard, E. (2006) A novel role for Xist RNA in the
formation of a repressive nuclear compartment into which genes are recruited when silenced. Genes Dev. 20, 2223–2237.
11. Chaumeil, J., Okamoto, I., Guggiari, M., and Heard, E. (2002) Integrated kinetics of X-chromosome
inactivation in differentiating embryonic stem cells. Cytogen. Gen. Res. 99, 75–84.
12. Chaumeil, J., Okamoto, I., and Heard, E. (2004) X-inactivation in mouse embryonic stem
cells: Analysis of histone modifications and transcriptional activity using immunofluores- cence and FISH. Methods Enzymol. 376, 405–419.
13. Chaumeil, J. (2005) Immunofluorescence–fluorescent in situ hybridization. Protocol for the
Epigenome Network of Excellence (http://www.epigenome-noe.net).
14. Spector, D.L., Goldman, R.D., and Leinwand, L.A. (1998) Cells: a laboratory manual. Cold
Spring Harbor Laboratory Press: Cold Spring Harbor, NY.
On the MEND A monthly patient's newsletter from MEND Physio Thank you for choosing MEND as YOUR physiotherapy clinic We firmly believe that clients will recover faster and more effectively if they are treated in a healthy environment, surrounded by healthy people, rather than a hospital setting where most people are ill!This is our first monthly newsletter and we hope that you will
Tel: 0161 476 3301 Mobile: 07903 162 041 How to Prepare for your Spray Tan: * Please wax or shave at least 24hrs prior to your Spray Tan - this allows enough time for your pores to close again. *Exfoliate your body 24hrs pior your Spray Tan using an exfoliating mitt or brush, paying particular attention to any very dry areas such as elbows, knees, feet etc. This will remove dead
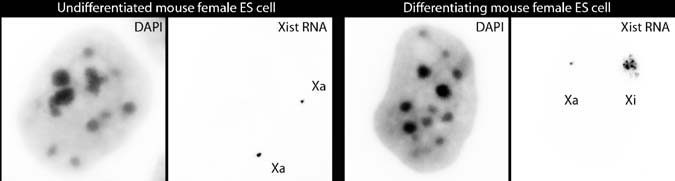
 18 Immunofluorescence, RNA FISH, and DNA FISH Combinations
Fig. 18.1 Xist RNA FISH in mouse female ES cells. In undifferentiated cells, Xist primary tran-
18 Immunofluorescence, RNA FISH, and DNA FISH Combinations
Fig. 18.1 Xist RNA FISH in mouse female ES cells. In undifferentiated cells, Xist primary tran-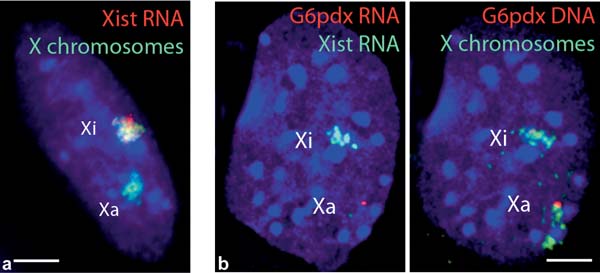
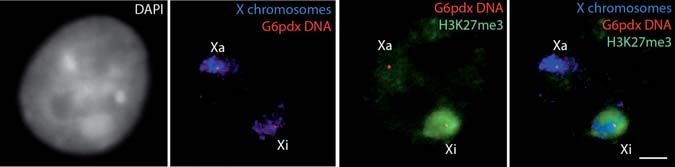 Fig. 18.3 Immunofluorescence combined with dual DNA FISH in differentiated female mouse ES
Fig. 18.3 Immunofluorescence combined with dual DNA FISH in differentiated female mouse ES