He encontrado que alguna farmacia puede tener existencias limitadas de ciertos medicamentos, mientras que otras pueden tener casi cualquier formato que se le ocurra y el habitual de dosis habitualidad apareció. En resumen, siempre se contiene el almacén de corroborar. Al mismo tiempo que el producto que más que gustaba ha resultado no estaba disponible en stock otro distinto por las Buenas costumbres también debe buscarse jefe no asн parezca. Por eso es importante disponer de un Plan B para actuar cuandod ello no ocurra.
Ventaja de tomar un genérico en lugar de Asix
Un genérico es más barato que el nombre de marca
Uno de los mayores incentivos para someterse al Dónde comprar Lasix genérico en lugar de pagar la marca es que usted puede obtener un ahorrando importantes Lasix genérico. Por lo tanto, un Lasix genérico es en general mucho más barato que el homólogo de marca, así que una denominación genérica se hace posible para las personas que usan este medicamento con frecuencia. Un ejemplo: La compra de lurosemida en lugar de Lasix es una considerable ahorro para el presupuesto mensual de medicamentos.
05-2215 860.868
Phase 1Trial of Gefitinib Plus Sirolimus in Adults with RecurrentMalignant Glioma
David A. Reardon,2,3 Jennifer A. Quinn,2,6 James J. Vredenburgh,2,6 Sridharan Gururangan,2,3Allan H. Friedman,2 Annick Desjardins,6 Sith Sathornsumetee,6 James E. Herndon II,7 Jeannette M. Dowell,7Roger E. McLendon,4 James M. Provenzale,5 John H. Sampson,2 Robert P. Smith,1Alan J. Swaisland,1Judith S. Ochs,1Peggy Lyons,2 Sandy Tourt-Uhlig,2 Darell D. Bigner,4 Henry S. Friedman,2,3and Jeremy N. Rich2,6
Purpose: To determine the maximum tolerated dose (MTD) and dose-limiting toxicity (DLT) ofgefitinib, a receptor tyrosine kinase inhibitor of the epidermal growth factor receptor, plussirolimus, an inhibitor of the mammalian target of rapamycin, among patients with recurrentmalignant glioma. Patients and Methods: Gefitinib and sirolimus were administered on a continuous daily dosingschedule at dose levels that were escalated in successive cohorts of malignant glioma patients atany recurrence who were stratified based on concurrent use of CYP3A-inducing anticonvulsants[enzyme-inducing antiepilepticdrugs, (EIAED)]. Pharmacokineticand archival tumor biomarkerdata were also assessed. Results: Thirty-four patients with progressive disease after prior radiation therapy and chemo-therapy were enrolled, including 29 (85%) with glioblastoma multiforme and 5 (15%) with ana-plasticglioma. The MTD was 500 mg of gefitinib plus 5 mg of sirolimus for patients not onEIAEDs and 1,000 mg of gefitinib plus 10 mg of sirolimus for patients on EIAEDs. DLTs includedmucositis, diarrhea, rash, thrombocytopenia, and hypertriglyceridemia. Gefitinib exposure wasnot affected by sirolimus administration but was significantly lowered by concurrent EIAED use. Two patients (6%) achieved a partial radiographic response, and 13 patients (38%) achievedstable disease. Conclusion:We show that gefitinib plus sirolimus can be safely coadministered on a continuous,daily dosing schedule, and established the recommended dose level of these agents incombination for future phase 2 clinical trials.
Traditional cytotoxic therapies, including external beam
Signal transduction pathways, associated with tumor cell
radiotherapy (X-ray therapy) and chemotherapy, provide a
proliferation, migration, angiogenesis, and survival, provide
modest survival advantage for some patients with newly
multiple potential therapeutic targets currently being evaluated
diagnosed glioblastoma multiforme (1). Salvage therapies are
in oncology. Aberrant signaling of the phosphatidylinositol 3V-
ineffective (2), and nearly all glioblastoma multiforme patients
kinase (PI3K) pathway occurs frequently in glioblastoma
die within 1 to 2 years of diagnosis. Innovative, more effective
multiforme (3) and is associated with poor response to
treatments are desperately needed for this patient population.
conventional cytotoxic therapy (4). Several molecular mecha-nisms have been linked to PI3K pathway signaling, includingactivation of upstream growth factor receptors, such as theepidermal growth factor receptor (EGFR), or loss of function
Authors’ Affiliations: 1AstraZeneca Pharmaceuticals, Wilmington, Delaware;
of the PTEN tumor suppressor gene, which normally
Departments of 2Surgery, 3Pediatrics, 4Pathology, 5Radiology, 6Medicine, and
antagonizes PI3K (5). We recently reported results of a clinical
7Cancer Center Biostatistics, Duke University Medical Center, Durham, North
trial with gefitinib, a novel low molecular weight, EGFR
CarolinaReceived 10/10/05; revised 11/6/05; accepted 11/11/05.
tyrosine kinase inhibitor (TKI), in recurrent glioblastoma
Grant support: NIH grants NS20023 and CA11898, NIH/General Clinical
multiforme patients. Although 9 of 53 patients (17%)
Research Center Program/National Center for Research Resources grant MO1 RR
remained progression-free for at least 6 months, the majority
30, and National Cancer Institute Specialized Programs of Research Excellence
of patients suffered early disease recurrence (6). Similar,
modest antitumor activity has recently been reported among
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby marked advertisement in accordance
recurrent glioblastoma multiforme patients treated with
with 18 U.S.C. Section 1734 solely to indicate this fact.
erlotinib, another EGFR TKI (7). Several possible factors may
Requests for reprints: David A. Reardon, The Preston Robert Tisch Brain Tumor
limit the clinical benefit associated with EGFR TKIs, including
Center at Duke, Duke University Medical Center, Box 3624, Durham, NC 27710.
compensatory activation of either downstream pathway
Phone: 919-668-2650; Fax: 919-668-2485; E-mail: [email protected].
components or alternative mitogenic/survival pathways, as
F 2006 American Association for Cancer Research. doi:10.1158/1078-0432.CCR-05-2215
well as molecular resistance mechanisms (8).
Clin Cancer Res 2006;12(3) February 1, 2006
Gefitinib Plus Sirolimus for Malignant Glioma
We recently showed in preclinical studies that the antitumor
activity of an EGFR TKI can be enhanced by combination with
Gefitinib and sirolimus were orally administered on a continuous
an inhibitor of the mammalian target of rapamycin (mTOR;
daily dosing schedule of 28-day cycles (Table 1). For all patients except
ref. 9). mTOR, a downstream target of the PI3K pathway, is a
for those who underwent pharmacokinetic sampling, a loading dose of
central regulator of several essential cellular processes in both
commercially available sirolimus was administered on the first day of
normal and neoplastic cells including nutrient metabolism,
cycle 1 followed by a continuous daily maintenance dose. Gefitinib,provided by AstraZeneca Pharmaceuticals (Wilmington, DE), was taken
cell cycle progression, and protein translation (10, 11).
concurrently with sirolimus. For patients who underwent pharmaco-
Although the clinical benefit of mTOR inhibitors for malig-
kinetic sampling, gefitinib was administered alone for days 1 to 7 of
nant glioma patients seems modest (12, 13), the antitumor
cycle 1. On day 8 of cycle 1, a loading dose of sirolimus was
activity of mTOR antagonists is enhanced by loss of PTEN
administered. Thereafter, gefitinib and sirolimus were administered
(14), which occurs commonly in glioblastoma multiforme
concurrently each day. Patients received up to 12 cycles unless
(15 – 18). Thus, possible mechanisms of EGFR TKI resistance
unacceptable toxicity or tumor progression occurred.
may be preferentially targeted by mTOR antagonists. To extend
Gefitinib metabolism is significantly enhanced by concurrent use of
this hypothesis to the development of a novel therapeutic
CYP3A-inducing antiepileptic drugs (EIAED), including phenytoin,
approach for malignant glioma patients, we conducted the
carbamazepine, phenobarbital, oxcarbazapine, and primidone (19).
current phase I study to determine the maximum tolerated
Therefore, patients were accrued independently into two separate strata:stratum A, patients not on EIAEDs; stratum B, patients on EIAEDs. A
dose (MTD) of gefitinib plus sirolimus, an mTOR antagonist,
‘‘3+3’’ phase I dose escalation design was employed to determine the
in patients with recurrent malignant glioma. Our report
MTD for each stratum. Intrapatient dose escalation was not permitted.
describes the first study of a combinatorial regimen of
The dose level was escalated in successive cohorts of three patients as long
molecularly targeted agents in the treatment of recurrent
as DLT did not occur. If one instance of DLT was observed among the
malignant glioma patients and specifically includes a combi-
initial three evaluable patients, three additional patients were treated at
natorial regimen designed to simultaneously inhibit key
that dose level. Dose escalation continued as long as no episodes of DLT
upstream and downstream mediators of PI3K signaling.
occurred in the additional three patients. If two instances of DLT wereobserved at a dose level, the MTD was surpassed and a total of six patientswere treated at the previous level. The MTD was defined as the highest
dose causing DLT in no more than one of six patients.
DLT was defined as any of the following toxicities that occurred
during the first cycle of therapy: grade 4 thrombocytopenia or
The primary objective was to define the MTD and dose-limiting
neutropenia lasting >4 days, grade z3 nonhematologic toxicities felt
toxicity (DLT) of gefitinib plus sirolimus in adults with recurrent
to be related to the study regimen excluding grade z3 nausea or emesis
malignant glioma. Secondary objectives included to further define the
for which inadequate medical therapy was administered, and >14 day
toxicity of this regimen, to obtain pharmacokinetic data, and to
delay in treatment due to related toxicity. Toxicities were graded
evaluate for antitumor activity relative to clinical, archival tumor
according to the National Cancer Institute’s Common Toxicity Criteria
biomarker, and pharmacokinetic measures.
version 3.0 and classified as related to the study regimen unless theywere attributable to either underlying tumor progression, a concurrent
medical condition or a concomitant medication.
Patients were required to have histologically confirmed malignant
Before each cycle, patients underwent a physical examination and
glioma (glioblastoma multiforme, gliosarcoma, anaplastic astrocyto-
full chemistry panel, including fasting cholesterol and triglycerides. A
ma, anaplastic oligodendroglioma, or anaplastic oligoastrocytoma)
complete blood count with differential was obtained weekly. In
that was radiographically progressive following prior radiation or
addition, before cycle 1, a urinalysis was obtained in all patients, and
chemotherapy. Additional enrollment criteria included age at least 18
h-human chorionic gonadotropin was obtained in women with
years, Karnofsky performance status z70%, stable corticosteroid dose
for at least 1 week before therapy initiation, hematocrit >29%; absolute
Response evaluation was done before each treatment cycle.
neutrophil count >1,000 cells/AL; platelet count >100,000 cells/AL,
Determination of overall response was based on radiographic change
serum creatinine and bilirubin <1.5 times the institutional upper limit
in tumor size as revealed by computed tomography or magnetic
of normal, serum aspartate aminotransferase <2.5 times the institu-tional upper limit of normal, and carbon monoxide diffusing capacityof >75% predicted. Patients were required to be at least 3 weeks from
prior surgical resection and to have recovered from all toxicitiesassociated with any prior therapy. All patients were informed of the
investigational nature of the study and provided informed consent as
approved by the Duke University Medical Center Institutional Review
Patients were excluded for any of the following: more that three
prior episodes of progressive disease, pregnancy or nursing, refusal to
use effective contraception if of reproductive potential, progressivedisease following prior treatment directed against either EGFR or
mTOR, and acute infection requiring i.v. antibiotics. In addition,
patients were not eligible if they received prior stereotactic radiosurgery,
radiation implants, or radiolabeled monoclonal antibody therapy, due
to the difficulty distinguishing progressive tumor from radionecrosis onmagnetic resonance imaging following such therapies. However,
*Stratum A: patients not on EIAEDs (phenytoin, phenobarbitol, carbamaze-
patients who had received these therapies were eligible if they had
pine, oxcarbazepine, and primidone).
either biopsy confirmation of recurrent tumor, or if they had a new or
progressive distant lesion on magnetic resonance imaging.
Clin Cancer Res 2006;12(3) February 1, 2006
resonance imaging and clinical criteria, including steroid requirement
Corp., Mountain View, CA). Total body clearance of drug from
and neurologic examination. Complete response was defined as the
plasma at steady state after an oral dose (CLss/F) was calculated as
disappearance of all enhancing or nonenhancing tumor from baseline
on consecutive scans at least 6 weeks apart, with the patient not
The paired t test was used to compare gefitinib alone AUCss (day 7)
receiving corticosteroids and neurologically stable or improved. Partial
to that with sirolimus (day 10) for each stratum and for each dose level.
response was defined as z50% reduction from baseline in the size
A two-sample t test was used to compare dose-normalized, gefitinib
(measured as the product of the largest perpendicular diameters) of
AUCss from day 7 between patients on strata A and B.
enhancing tumor maintained for at least 6 weeks, use of a stable or
A trough serum sirolimus level was measured after day 10 of cycle
reduced corticosteroid dose, and stable or improved neurologic exam.
1. Two-way ANOVA was used in a generalized linear model
Progressive disease was defined as >25% increase in size of enhancing
framework to examine the effect of dose and strata on blood levels
or nonenhancing tumor or any new tumor on magnetic resonance
of sirolimus. This analytic approach assumed measurement errors to
imaging scan or neurologic worsening of the patient without a
be normally distributed, and repeated measures of sirolimus levels
documented nonneurologic etiology while on a stable or increased
corticosteroid dose. Stable disease was defined as any other status notmeeting the criteria for complete response, partial response, and
progressive disease that was observable for more than one course of
Archival tumor samples from either initial diagnosis or after prior
therapy were analyzed for phospho-p44/42 mitogen-activated protein
Time to progression and overall survival, measured from the date
kinase (p-MAPK), p-S6 ribosomal protein, p-AKT, PTEN, and EGFR
cycle 1 began, were analyzed by the Kaplan-Meier method, including
using immunohistochemistry reagents and methods as described
95% confidence intervals (95% CI; refs. 20, 21).
below. Similarly, archival tumor samples were analyzed by fluorescencein situ hybridization (FISH) for EGFR and PTEN DNA locus copy
Dose modification and retreatment criteria
number using reagents and methods as described below. Primary
The criteria for retreatment consisted of the following: absolute
antibodies for immunohistochemical staining included rabbit mono-
neutrophil count > 1,000 cells/AL; platelets >100,000 cells/Al; serum
clonal p-MAPK (Thr202/Tyr204, clone E10), rabbit polyclonal p-S6
aspartate aminotransferase, total bilirubin, and creatinine <1.5 times
ribosomal protein (Ser235/Ser236), rabbit polyclonal p-AKT (Ser473; Cell
upper limit of normal and resolution of all related toxicities to grade V1
Signaling Technology, Boston, MA), and mouse monoclonal PTEN
except for rash, which was required to improve to grade V 2. For
(clone 6H2.1; Cascade Bioscience, Inc., Winchester, MA). The
patients who develop DLT regardless of treatment cycle, the study
EGFRpharmDx kit (DAKO Corp., Carpinteria, CA) was used for EGFR
regimen was reduced to the dose level below that on which the patient
was entered. Patients were removed from study for evidence of
Primary antibodies were used at the following dilutions and
progressive disease at any time after study initiation, grade 4
incubations: p-MAPK, 1:100 overnight at 4jC; p-S6 ribosomal protein,
nonhematologic toxicity, more than two dose reductions due to
1:100 for 1 hour at room temperature; p-AKT, 1:50 overnight at 4jC;
toxicity, dose reduction of gefitinib to <250 mg/d, noncompliance, or
and PTEN, 1:1000 overnight at 4jC. The EGFR antibody was provided
at a predetermined dilution, and immunohistochemistry was doneaccording to the Food and Drug Administration – approved manufac-
turer’s protocol for the DAKO EGFRpharmDx kit.
Antiemetic therapy with ondansetron and dexamethasone was
Immunohistochemistry. For all immunohistochemistry assays, 5-Am
permitted if needed. Loperamide was prescribed for diarrhea as
sections were cut from paraffin-embedded, formalin-fixed brain tissue,
previously described (22). Hematopoietic growth factors and blood
placed on silanized slides, deparaffinized with a series of xylenes,
products were administered as indicated for hematologic DLT or
cleared in a series of alcohols, and rehydrated. Endogenous peroxidase
hematologic toxicity that occurred after cycle 1. Lipid lowering agents
were permitted if prescribed before study enrollment, or for patients
Antigen retrieval was done by one of several methods. For
who developed either DLT or hyperlipidemia after cycle 1. Significant
p-S6 and p-AKT, a solution of 10 mmol/L EDTA was used in a
rash was treated with over-the-counter acne preparations, antihist-
decloaking chamber for 5 minutes at 120jC. For p-MAPK and PTEN, a
amines, and topical clindamycin and/or oral antibiotics (penicillins or
sodium citrate solution (pH 6.0) was used in a Black and Decker
Following antigen retrieval, slides were washed in TBS with 0.1%
Tween 20, and nonspecific protein binding was blocked with 5%
Venous blood samples (4 mL) were collected for gefitinib
normal goat serum for 15 minutes at room temperature. For p-AKT
pharmacokinetic studies from patients on days 7 and 10 of cycle
and p-S6, a 30-minute incubation with goat anti-rabbit secondary
1 before the daily dose and 1, 2, 4, 6, 8, and 24 hours after the
antibody was followed by detection with avidin-biotin complex Elite
daily dose. For each sample, plasma supernatants were separated by
kit (Vector Laboratories, Burlingame, CA). For PTEN, a 30-minute
centrifugation (1,000 Â g for 10 minutes at room temperature) and
incubation with goat anti-mouse supersensitive link was followed by
immediately frozen at À20jC. Plasma concentrations of gefitinib
detection with Super Sensitive Detection Kit (Biogenex, San Ramon,
were determined by high-pressure liquid chromatography with
CA). For MAPK, a 30-minute incubation with goat anti-rabbit
tandem mass spectrometry detection by the Drug Metabolism and
secondary antibody was followed by detection with the Multilink
Pharmacokinetics Department at AstraZeneca, Alderley Park, United
Detection kit (Biogenex, San Ramon, CA). Nuclear counterstaining
was done using Harris’ modified hematoxylin. The intensity of
Steady-state plasma drug concentrations were used to provide a
cytoplasmic/membranous staining detected by immunohistochemistry
measure of exposure and the pharmacokinetic variables. Maximum
was scored on a scale of 0 to 4+, and the distribution was defined as
steady-state plasma gefitinib concentration during the dosing interval
the percentage of cells with any level of expression. Immunohisto-
(Css,max), the time to reach maximum gefitinib concentration (Tmax),
chemical staining was defined as ‘‘high’’ for tumors expressing 2 to 4+
and the minimum concentration during the dosing interval at steady
intensity in z25% of tumor cells and as ‘‘low’’ for tumors expressing
state (Css,min), defined as the concentration at 24 hours after dose on
either 0 to 1+ staining in any percentage of tumor cells or 2 to 4+
each sample day, were obtained directly from the data. The area
intensity in <25% of tumor cells (3).
under the concentration versus time curve at steady state (AUCss) was
Dual-color FISH was done on formalin-fixed, paraffin-
calculated by the linear trapezoidal rule using WinNonlin (Pharsight
embedded tissue specimens using the EGFR/CEP 7, CEP 10/CEP 2
Clin Cancer Res 2006;12(3) February 1, 2006
Gefitinib Plus Sirolimus for Malignant Glioma
(Vysis, Downers Grove, IL), and CEP 10/PTEN (Human BAC CITB
Slides were viewed using an Olympus BX-60 fluorescent microscope.
library clone 265N13, Research Genetics, Huntsville, AL) probe
The number of green and orange signals was enumerated in 100 intact,
combinations (using three separate slides) for each patient sample.
nonoverlapping nuclei per slide. With regard to chromosomal gain, the
CEP 2 was chosen as an internal control for the loss of chromosome 10
cutoff value was set at 20%, meaning that >20% of the enumerated
(23). The EGFR probe does not discriminate between wild-type EGFR
nuclei must show more than two copies of the respective probe. For
chromosomal loss, the cutoff value was set at 30% for definitive loss
Paraffin sections were cut at 5 Am onto silanized slides. Control and
and 20% to 30% for indeterminate loss. EGFR gene amplification was
patient slides were baked overnight at 56jC. Formalin-fixed, paraffin-
defined as an EGFR/chromosome 7 centromere ratio of >2.0. Definitive
embedded control cell lines, showing the locus of interest, were used as
PTEN loss was defined as tumors in which z30% of nuclei exhibited
less than two copies of the PTEN locus and two copies of CEP 2
Slides were deparaffinized, pretreated with 0.2 N HCl at room
control. Indeterminate PTEN loss refers to tumors in which 20% to
temperature for 20 minutes, then washed in deionized water and 2Â
30% of enumerated nuclei had less than two copies of the PTEN locus
SSC for 3 minutes each. They were then placed in Pretreatment
Solution (Vysis) at 80jC for 30 minutes and washed with two changesof 2Â SSC for 5 minutes each. Sections were subjected to digestion with
protease at 37jC for 20 to 23 minutes. Slides were washed in twochanges of 2Â SSC for 5 minutes each and air-dried, then were
denatured in a 70% formamide/2Â SSC solution at 72jC for 5 minutes
malignant glioma were enrolled at the Duke University
and immediately dehydrated in 70%, 85%, and 100% ethanol for 1
Medical Center between August 2004 and February 2005
minute each. Subsequently, the probe was denatured at 75jC for 5
(Table 2). Twenty-nine patients had glioblastoma multiforme
minutes. Fluoresceinated probe was applied to each slide, sealed with
(85%) and 5 (15%) had anaplastic astrocytoma. Fifteen
rubber cement, and then placed in a humidified chamber at 37jC foran overnight incubation. After overnight incubation, slides were then
patients (44%) were not on EIAEDs (stratum A) and 19
washed in 2Â SSC/0.3% NP40 at room temperature and then at 72jC
(56%) were on EIAEDs (stratum B). Patient characteristics did
for 2 minutes. 4V,6-Diamidino-2-phenylindole counterstain and a
not differ substantially based on EIAED status. Twenty-three
coverslip were applied to the hybridization area.
patients (68%) were male. The median age was 49.9 years
Abbreviations: GBM, glioblastoma multiforme; GS, gliosarcoma; AA, anaplastic astrocytoma; KPS, Karnofsky performance status; XRT, X-ray therapy.
*EIAEDs: phenytoin, carbamazepine, phenobarbitol, oxcarbazepine, and primidone.
Clin Cancer Res 2006;12(3) February 1, 2006
(range, 32.8-76.8 years). All patients had a Karnofskyperformance status of at least 70%.
All patients had received prior X-ray therapy and chemo-
therapy. The median number of prior chemotherapeutic agents
administered per patient was 2 (range, 1-6). The median
number of prior episodes of progressive disease per patient was
2 (range, 1-3). The median time from original diagnosis to
initiation of study treatment was 29.8 weeks (range, 7.3-248.0
As of September 15, 2005, five patients continue to receive
treatment on study with stable disease. Twenty-one patients
and type of DLT observed at each dose level per stratum. Onegroup A patient developed fulminant progressive disease
*Stratum A: patients not on EIAEDs (phenytoin, phenobarbitol, carbamaze-
and discontinued study treatment after <2 weeks of cycle 1.
pine, oxcarbazepine, and primidone).
Although this patient did not experience a DLT, they were
cOne additional patient was treated at the MTD (dose level1) for stratum A for
replaced in the cohort for MTD determination. However, this
bOne patient treated at dose level 2 of stratum A was not eligible for MTD
patient was included in overall toxicity assessment. One
determination due to fulminant progressive disease.
additional patient was added at dose level one for stratum A
xOne patient treated at dose level 2 of stratum A and two patients treated at
and provided additional safety and pharmacokinetic data.
dose level 3 of stratum B interrupted dosing during cycle 1and were replaced
Three patients (one in dose level 2 of stratum A and two in
dose level 3 of stratum B) decreased or interrupted dosing
Stratum B: patients on EIAEDs (phenytoin, phenobarbitol, carbamazepine,
during cycle 1 due to miscommunication or noncompliancewith administration guidelines. Although these patients wereassessable for DLT, they were not assessable for dose
Limited sampling from two stratum B patients treated on
escalation within each cohort and were therefore replaced.
dose level 3 was available and is therefore not included.
For stratum A, one of seven patients treated at dose level 1
Comparison of day 7 (gefitinib alone) and day 10 (gefitinib
experienced DLT (grade 3 mucositis), whereas two of seven
plus sirolimus) measures revealed that sirolimus, a known
patients treated at dose level 2 experienced DLT, including one
substrate for CYP3A4, did not affect gefitinib metabolism.
patient with grade 3 thrombocytopenia that required >2 weeks
However, gefitinib exposure was significantly reduced by
to resolve to retreatment criteria, and one patient with grade 3
concurrent EIAED use. Specifically, the dose-normalized,
rash and mucositis. For stratum B, none of the patients
geometric mean of AUCss for patients not on (stratum A) and
experienced DLT at dose level 1. However, one of six patients
for those on EIAEDs (stratum B) were 19.9 and 7.91 ng h/mL,
treated at dose level 2 developed dose-limiting hypertriglycer-
idemia, whereas two of eight patients treated at dose level 3
Trough sirolimus data was available on 23 patients, including
developed DLT, including one patient with grade 3 diarrhea
12 patients from stratum A and 11 patients from stratum B. The
and one patient with grade 3 mucositis.
mean trough sirolimus level for patients treated with 5 mg/d
Non-DLT. One hundred courses of gefitinib plus sirolimus
(7.0) was significantly less than that of patients treated with 10
have been administered to date, including 44 courses to
mg/d (16.7; P < 0.0001); however, trough sirolimus levels did
patients on stratum A and 56 courses to patients on stratum B.
not differ based on stratum (P = 0.136).
Table 4 summarizes the most frequent toxicities stratified by
Archival tumor biomarker analysis. Archival tumor material
toxicity grade and treatment stratum.
was available for 14 patients (Table 6). FISH analysis revealed
Diarrhea, mucositis, and rash were the most common
that 6 patients had EGFR amplification (43%) and 7 patients
toxicities as expected. Hematologic toxicity and fasting
had evidence of PTEN loss (50%). ‘‘High’’ levels of EGFR,
cholesterol or triglyceride elevations were also noted, primar-
p-S6, p-MAPK, and p-AKT were detected by immunohisto-
ily as low-grade events, and also as infrequent grade 3 or 4
chemistry in 90% (9 of 10), 60% (6 of 10), 60% (6 of 10),
events. Grade 1 or 2 infections, most frequently involving the
and 90% (9 of 10) of assessable patients, respectively. A
skin and nailbeds, were also noted. Two serious infections
good correlation was observed between EGFR amplification
occurred among patients on study and included episodes of
detected by FISH and EGFR expression by immunohistochem-
grade 3 and 4 pneumonia, respectively. The episode of grade 4
istry. All four tumors with EGFR amplification by FISH
pneumonia was most likely due to aspiration following a
showed 3 to 4+ EGFR expression in 90% to 100% of cells by
seizure. Both events resolved with i.v. antibiotics and
immunohistochemistry. Of note, three of four tumors with
hospitalization. One patient, treated with gefitinib plus
evidence of PTEN loss by FISH had elevated p-AKT expression
sirolimus for 7 months, developed disseminated Aspergillus
f2 months following study discontinuation while receiving
Outcome. All 34 patients were evaluable for response. Two
an alternative, salvage therapy. Of note, there were no grade 5
patients achieved a partial radiographic response, including
one patient treated at dose level 1 on stratum A (Fig. 1) and
Pharmacokinetic analyses. Ten patients from stratum A and
another patient treated at dose level 3 on stratum B. Thirteen
nine patients from stratum B underwent plasma gefitinib
patients (38%) achieved stable disease, including 7 patients on
stratum A (47%) and 6 patients on stratum B (32%). By
Clin Cancer Res 2006;12(3) February 1, 2006
Gefitinib Plus Sirolimus for Malignant Glioma
Table 4. Most frequent toxicities stratified by grade and patient stratum
Abbreviations: AST, aspartate aminotransferase; ALT, alanine aminotransferase. *Stratum A: patients not on EIAEDs (phenytoin, carbamazepine, oxcarbazepine, phenobarbitol, and primidone).
cStratum B: patients on EIAEDs (phenytoin, carbamazepine, oxcarbazepine, phenobarbitol, and primidone). bIncludes only grade 1and 2 events that occurred at a minimum of three times.
xIncludes all grade 3 and 4 events including those reported as DLT.
kCumulative number of events observed among all cycles of therapy.
{Percentage of all patients in stratum.
histology, 12 patients with glioblastoma multiforme (41%)
weeks). Analysis of possible associations among clinical,
and 1 with recurrent anaplastic glioma (20%) achieved stable
pharmacokinetic, and archival tumor biomarker variables with
outcome was limited by study accrual and the dose escalation
With a median follow-up of 35.2 weeks (95% CI, 27.7-42.4),
the median progression-free disease (PFS) and 6-month PFSrate for all patients were 8.2 weeks (95% CI, 7.5-18.6 weeks)
and 23.5% (95% CI, 12.8-43.1%), respectively. Median PFSand 6-month PFS did not differ significantly by histology or
The rationale for the study regimen of gefitinib plus sirolimus
EIAED status. Among patients who achieved at least stable
is that simultaneously targeting key upstream and downstream
disease, the median PFS was 27.4 weeks (95% CI, 18.7-30.6
mediators of PI3K signaling may produce greater antitumor
Table 5. Gefitinib pharmacokinetic variables
Abbreviations: Gmean, geometricmean; CV%, coefficient of variation.
*EIAEADs: phenytoin, carbamazepine, oxcarbazepine, phenobarbitol, primidone.
Clin Cancer Res 2006;12(3) February 1, 2006
activity than that achieved when either mediator is targeted
downstream signaling mediators or alternative signaling
separately. Results of preclinical studies confirm that such
pathways may provide compensatory proliferative and survival
combinatorial regimens are capable of synergistic antitumor
activity (9, 24 – 27). Furthermore, such combinatorial regi-
Our phase I study achieved its primary objective of
mens may be less vulnerable to resistance mechanisms against
establishing the MTD of a continuous daily dosing regimen
targeted therapeutics. Although current understanding of such
of gefitinib plus sirolimus for patients with recurrent malignant
resistance mechanisms is limited (28), insights can be gained
glioma. Specifically, the MTD is 500 mg of gefitinib plus 5 mg
from both clinical and preclinical studies. For example, the
of sirolimus for patients not on EIAEDs, and 1,000 mg of
majority of patients exhibit resistance to EGFR TKIs, although
gefitinib plus 10 mg of sirolimus for those on EIAEDs.
most glioblastoma multiforme tumors express EGFR, suggest-
Furthermore, we show that these agents can be safely
ing that compensatory mechanisms can overcome EGFR
combined at doses used in monotherapy dosing schedules
inhibition. One such compensatory mechanism may be
(6, 41, 42). There were no unexpected toxicities and the
increased activity of additional growth factor receptors.
spectrum of observed toxicities, including DLTs, was similar to
Growth factors reported to be overexpressed in malignant
those previously reported in monotherapy studies (6, 12, 13,
glioma include platelet-derived growth factor receptor (29,
41, 42). Although not observed among enrolled patients,
30), vascular endothelial growth factor (31), fibroblast growth
opportunistic infections pose an appropriate concern with this
factor receptor (32), and insulin-like growth factor-I receptor
regimen due to the immunosuppressive activity of sirolimus,
(33). Furthermore, some EGFR-resistant tumors exhibit
particularly because malignant glioma patients are inherently
activation of alternative growth factor receptor pathways,
immunocompromised (43, 44) and are frequently on immu-
suggesting that either tumor mitogenesis or induction of
angiogenesis may act to compensate for EGFR signaling loss
Secondary objectives of this study included the evaluation of
(26, 33 – 36). EGFR TKI resistance has also been associated
pharmacokinetic end points, the assessment of biomarkers
with increased activity of intracellular mediators. For example,
from archival tumor specimens of enrolled patients, and the
loss of the PTEN tumor suppressor, which constitutively
determination of evidence of antitumor activity. Our phar-
activates AKT (36, 37), is linked to resistance to EGFR-based
macokinetic studies confirmed that gefitinib exposure is
therapies (25, 38 – 40). In a recent glioblastoma multiforme
significantly affected by concurrent EIAED use and provided
trial, elevated levels of p-AKT correlated with erlotinib
reassurance that sirolimus does not affect gefitinib metabolism.
resistance (7). These data suggest that in response to upstream
The analysis of our immunohistochemistry and FISH
growth factor TKI therapy, enhanced activity of either
findings was limited by specimen availability and the dose
Table 6. Archival tumor biomarker analysis
Abbreviations: ND, not done; PR: partial response; SD: stable disease; PD: progressive disease; TTP: time to progression; I, intensity; D, distribution. *FISH: amplified EGFR : EGFR/chromosome 7 ratio >2.0; polysomy EGFR : EGFR/chromosome 7 ratio 1-2; PTEN loss: <2 copies PTEN with 2 copies of CEP 2 control in
z30% nuclei; PTEN indeterminate: <2 copies PTEN with 2 c opies of CEP 2 c ontrol in 20% to 30% nuc lei.
cImmunohistochemistry: wild-type EGFR, p-S6 ribosomal protein, p-p44/42 MAPK, p-AKT. I, most common staining pattern present overall (0+, no staining; 1+,
minimal cytoplasmic/membraneous staining; 2+, mild cytoplasmic/membraneous staining; 3+, moderate cytoplasmic/membraneous staining; 4+, strong cytoplasmic/membraneous staining). D, percent positive cells; focal refers to heterogeneous, regional, or sporadic staining of <25% of evaluated tumor cells.
bStratum A: no EIAEDs; stratum B, on EIAEDs.
Clin Cancer Res 2006;12(3) February 1, 2006
Gefitinib Plus Sirolimus for Malignant Glioma
dynamic measures to assess the study regimen’s effect onintratumoral PI3K and mTOR signaling was not assessed. Therefore, confirmation that either study agent was success-fully delivered at dose levels required to inhibit the intendedintracellular target was not obtained. Ongoing and plannedclinical trials with EGFR and mTOR inhibitors that incorpo-rate pharmacodynamic evaluations of tumor cell targets mayclarify this critical issue. Finally, it is possible that suppress-ing both EGFR and mTOR may not be sufficient to effectivelytreat some glioblastoma multiforme tumors due to aberrantactivation of alternative downstream PI3K mediators or othergrowth factor/survival pathways. The identification of severalsignal transduction pathways commonly altered in malignantglioma suggests that targeting pathways in parallel may alsocontribute to effective therapeutic synergy.
In conclusion, we report the first clinical trial incorporating
a combinatorial regimen of signal transduction inhibitors formalignant glioma patients. In addition to establishing theMTD of this regimen, we confirm that therapeutics targetingEGFR and mTOR can be safely coadministered to malignantglioma patients. Phase 2 trials to evaluate the antitumoractivity of EGFR and mTOR targeting regimens are under wayfor recurrent malignant glioma patients. Combinatorialregimens, including those designed to simultaneously target
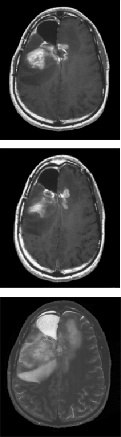
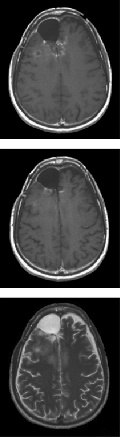
Fig.1. Partial response to gefitinib plus sirolimus. Top and middle, axial T1-weighed
key upstream and downstream signaling mediators, represent
sequences following gadolinium administration; bottom,T2-weighed sequences. After one cycle of treatment, a marked reduction of both contrast-enhancing tumor
an important advance in the evaluation of targeted therapeu-
and associated edema was observed. The radiographic response, accompanied by
tics for cancer patients. The therapeutic potential of such
marked clinical improvement, was maintained for 4 months, at which point aprogressive tumor developed.
combinatorial approaches for future studies is noteworthy butcritically hinges on the comprehensive integration of clinical,pretreatment tumor biomarker, pharmacokinetic and intra-
escalation design of this study. Furthermore, tumor samples
evaluated in our trial were obtained at either initial diagnosisor after prior therapy and therefore may not have reflected theactual molecular genetic profile of the tumor at study entry.
Nonetheless, the potential of such assays to prospectively
identify appropriate cohorts of malignant glioma patientsfor treatment with selected targeted therapeutics was recently
shown (7). In this analysis, patients with archival tumorsamples showing p-AKT and EGFR amplification had a
significantly greater likelihood of response to the EGFR TKI
The rate of radiographic response on the current study was
comparable with that observed among glioblastoma multi-
forme patients treated with temozolomide at first recurrence
(45). However, PFS on the current study was similar to that
achieved on our prior phase II study with gefitinib alone (6).
Although the assessment of antitumor activity is limited in
any phase I study, several additional factors may have
affected our study’s outcome. First, patients were heavily
pretreated, having enrolled following treatment with a
median of two prior chemotherapy agents (range, 1-6) and
a median of two prior recurrences (range, 1-3). Second,
nearly all patients on the current study had bulky measurable
tumor, whereas only 11 of 53 patients (21%) enrolled onour prior phase 2 study had measurable tumor (6). Third,
Abbreviations: PR, partial response; SD, stable disease; PD, progressive
EGFRvIII expression, which was unable to be assessed in the
current study due to technical factors with the EGFRvIII
*% Patients with an evaluable assay.
immunohistochemistry assay, may have also affected re-
cAmplified: EGFR/chromosome 7 centromere ratio of >2.0.
sponse (7, 46). Fourth, our pharmacokinetic studies confirm
bLoss: 30% of nuclei with definitive loss and 20-30% with indeterminate loss.
x High, 1-4+ intensity in z25% of tumor cells; low, either no staining or 1-4+
that concurrent use of EIAEDs markedly diminish gefitinib
intensity in <25% of tumor cells.
exposure. Finally, and perhaps most importantly, pharmaco-
Clin Cancer Res 2006;12(3) February 1, 2006
References1. Stupp R, Mason WP, van den Bent MJ, et al. Radio-
tification of a candidate tumour suppressor gene,
WF. Immunolocalization of basic fibroblast growth
therapy plus concomitant and adjuvant temozolomide
MMAC1, at chromosome 10q23.3 that is mutated
factor to the microvasculature of human brain tumors.
for glioblastoma. N Engl J Med 2005;352:987 ^ 96.
in multiple advanced cancers. Nat Genet 1997;15:
2. Wong ET, Hess KR, Gleason MJ, et al. Outcomes and
33. Chakravarti A, Loeffler JS, Dyson NJ. Insulin-like
prognosticfactors in recurrent glioma patients enrolled
19. Swaisland H, Smith R, Farebrother J, Laight A. The
growth factor receptor I mediates resistance to
onto phase II clinical trials. J Clin Oncol 1999;17:
effect of the induction and inhibition of CYP3A4 on
anti-epidermal growth factor receptor therapy in pri-
the pharmacokinetics of single oral does of ZD1839
mary human glioblastoma cells through continued
3. Choe G, Horvath S, Cloughesy TF, et al. Analysis of
(‘Iressa’), a selective epidermal growth factor receptor
activation of phosphoinositide 3-kinase signaling.
the phosphatidylinositol 3V-kinase signaling pathway
tyrosine kinase inhibitor (EGFR-TkI), in healthy male
in glioblastoma patients in vivo. Cancer Res 2003;63:
volunteers. In: ProcAm SocClin Oncol. Orlando, FL;
34. Natale R, Shak S, Aronson N, et al. Quantitative
gene expression in non-small cell lung cancer from
4. Chakravarti A, Zhai G, Suzuki Y, et al. The prognostic
20. Brookmeyer R, Crowley J. A confidence interval
paraffin-embedded tissue specimens: Predicting re-
significance of phosphatidylinositol 3-kinase pathway
for the median survival time. Biometrics 1982;38:
sponse to gefitinib, an EGFR kinase inhibitor. In:
activation in human gliomas. J Clin Oncol 2004;22:
ProcAm SocClin Oncol, Chicago, Illinois, May 31-
21. Klein JP. Small sample moments of some estimators
5. Vivanco I, Sawyers CL. The phosphatidylinositol
of the variance of the Kaplan-Meier and Nelson-Aalen
35. Haas-Kogan D, Shalev N, Wong M, et al. Protein
3-kinase AKT pathway in human cancer. Nat Rev Can-
estimators. Scand J Stat 1991;18:333 ^ 40.
kinase B (PKB/Akt) activity is elevated in glioblastoma
22. Friedman HS, Petros WP, Friedman AH, et al. Iri-
cells due to mutation of the tumor suppressor PTEN/
6. Rich JN, Reardon DA, Peery T, et al. Phase II trial of
notecan therapy in adults with recurrent or pro-
gefitinib in recurrent glioblastoma. J Clin Oncol 2004;
gressive malignant glioma. J Clin Oncol 1999;17:
36. Davies MA, LuY, SanoT, et al. Adenoviral transgene
expression of MMAC/PTEN in human glioma cells
7. Haas-Kogan DA, Prados MD,TihanT, et al. Epidermal
23. Wiltshire RN, Herndon JE II, Lloyd A, et al. Compar-
inhibits Akt activation and induces anoikis. Cancer
growth factor receptor, protein kinase B/Akt, and glio-
ative genomichybridization analysis of astrocytomas:
ma response to erlotinib. J Natl Cancer Inst 2005;97:
prognosticand diagnosticimplications. J Mol Diagn
37. Li B, Chang CM, Yuan M, McKenna WG, Shu HK.
Resistance to small molecule inhibitors of epidermal
8. Camp ER, Summy J, Bauer TW, et al. Molecular
24. Amador ML, Maitra A, Gruenwald V, Peralba JM,
growth factor receptor in malignant gliomas. Cancer
mechanisms of resistance to therapies targeting the
Hidalgo M. Determinants of resistance to OSI-774 in
epidermal growth factor receptor. Clin Cancer Res
billiary tract carcinoma cell lines. In: Proc Am Soc
38. She QB, Solit D, Basso A, Moasser MM. Resis-
Clin Oncol, Chicago IL, May 31-June 3, 2003 2003,
tance to gefitinib in PTEN-null HER-overexpressing
9. Goudar RK, Shi Q, Hjelmeland MD, et al. Combi-
tumor cells can be overcome through restoration of
nation therapy of inhibitors of epidermal growth
25. Fan QW, Specht KM, Zhang C, et al. Combinatorial
PTEN function or pharmacologic modulation of con-
factor receptor/vascular endothelial growth factor
efficacy achieved through two-point blockade within
stitutive phosphatidylinositol 3V-kinase/Akt pathway
receptor 2 (AEE788) and the mammalian target of
a signaling pathway-a chemical genetic approach.
signaling. Clin Cancer Res 2003;9:4340 ^ 6.
rapamycin (RAD001) offers improved glioblastoma
39. Bianco R, Shin I, Ritter CA, et al. Loss of PTEN/
tumor growth inhibition. Mol Cancer Ther 2005;4:
26. Perez-Soler R, Ling Y, Lia M, et al. Molecular mech-
MMAC1/TEP in EGF receptor-expressing tumor cells
anisms of resistance to the HER1/EGFR tyrosine kinase
counteracts the antitumor action of EGFR tyrosine
10. Bjornsti MA, Houghton PJ. The TOR pathway: a
inhibitor erlotinib HCI in human cell lines. In: Proc Am
kinase inhibitors. Oncogene 2003;22:2812 ^ 22.
target for cancer therapy. Nat Rev Cancer 2004;4:
Soc Clin Oncol, Chicago IL, May 31-June 3, 2003
40. Kokubo Y, Gemma A, Noro R, et al. Reduction of
PTEN protein and loss of epidermal growth factor re-
11. SchmelzleT, Hall MN. TOR, a central controller of cell
27. Rao R, Sarkaria J, Frederick L, Erlichman C, CD J.
ceptor gene mutation in lung cancer with natural resis-
Synergisticinhibition of glioma cell growth upon com-
tance to gefitinib (IRESSA). Br J Cancer 2005;92:
12. Chang SM, Kuhn J, Wen P, et al. Phase I/pharma-
bination therapy with the mTOR inhibitor, rapamycin,
cokinetic study of CCI-779 in patients with recurrent
and the epidermal growth factor receptor inhibitor,
41. Giaccone G. Epidermal growth factor receptor
malignant glioma on enzyme-inducing antiepileptic
EKI-785. In: American Association of Cancer Research
inhibitors in the treatment of non-small-cell lung can-
drugs. Invest New Drugs 2004;22:427 ^ 35.
Annual Meeting, Toronto, Ontario, Canada, April 5 ^ 9,
cer. J Clin Oncol 2005;23:3235 ^ 42.
13. Galanis E, BucknerJC, Maurer MJ, et al. Phase II trial
42. Kuypers DR. Benefit-risk assessment of siroli-
of Temsirolimus (CCI-779) in recurrent glioblastoma
28. Viloria-Petit AM, Kerbel RS. Acquired resistance
mus in renal transplantation. Drug Saf 2005;28:
multiforme: North Central Cancer Treatment Group.
to EGFR inhibitors : mechanisms and prevention
strategies. Int J Radiat Oncol Biol Phys 2004;58:
43. Mahaley MS, Jr., Brooks WH, Roszman TL, et al.
14. Neshat MS, Mellinghoff IK, Tran C, et al. Enhanced
Immunobiology of primary intracranial tumors. Part 1:
sensitivity of PTEN-deficient tumors to inhibition of
29. Fleming TP, Saxena A, Clark WC, et al. Amplifica-
studies of the cellular and humoral general immune
FRAP/mTOR. ProcNatl Acad Sci U S A 2001;98:
tion and/or overexpression of platelet-derived growth
competence of brain-tumor patients. J Neurosurg
factor receptors and epidermal growth factor recep-
15. Duerr EM, Rollbrocker B, Hayashi Y, et al. PTEN
tor in human glial tumors. Cancer Res 1992;52:
44. RoszmanTL, Elliott LH, BrooksWH. Proliferative po-
mutations in gliomas and glioneuronal tumors. Onco-
tential of T-cell lymphocytes from gliomas. J Neuro-
30. Hermanson M, Funa K, Hartman M, et al. Platelet-
16. Li J, Yen C, Liaw D, et al. PTEN, a putative protein
derived growth factor and its receptors in human glio-
45. YungWK, Albright RE, Olson J, et al. A phase II study
tyrosine phosphatase gene mutated in human brain,
ma tissue: expression of messenger RNA and protein
of temozolomide vs. procarbazine in patients with
breast, and prostate cancer. Science 1997;275:
suggests the presence of autocrine and paracrine
glioblastoma multiforme at first relapse. Br J Cancer
17. Rasheed BK, Stenzel TT, McLendon RE, et al.
31. Takano S, Yoshii Y, Kondo S, et al. Concentration of
46. Learn CA, Hartzell TL, Wikstrand CJ, et al. Resis-
PTEN gene mutations are seen in high-grade but
vascular endothelial growth factor in the serum and
tance to tyrosine kinase inhibition by mutant epider-
not in low-grade gliomas. Cancer Res 1997;57:
tumor tissue of brain tumor patients. Cancer Res
mal growth factor receptor variant III contributes to
the neoplasticphenotype of glioblastoma multi-
18. Steck PA, Pershouse MA, Jasser SA, et al. Iden-
32. Brem S, Tsanaclis AM, Gately S, Gross JL, Herblin
forme. Clin Cancer Res 2004;10:3216 ^ 24.
Clin Cancer Res 2006;12(3) February 1, 2006
From Global Enclosure to Self Enclosure: Ten Years After – A Critique of the CBD and the “Bonn Guidelines” on Access and Benefit Sharing (ABS) Issue: Since 1994, the Convention on Biological Diversity (CBD) has been promising “benefit sharing” to Indigenous Peoples in return for access to biodiversity (i.e., bioprospecting). During these tenyears, Indigenous People
 Gefitinib Plus Sirolimus for Malignant Glioma
dynamic measures to assess the study regimen’s effect onintratumoral PI3K and mTOR signaling was not assessed.
Gefitinib Plus Sirolimus for Malignant Glioma
dynamic measures to assess the study regimen’s effect onintratumoral PI3K and mTOR signaling was not assessed.