He encontrado que alguna farmacia puede tener existencias limitadas de ciertos medicamentos, mientras que otras pueden tener casi cualquier formato que se le ocurra y el habitual de dosis habitualidad apareció. En resumen, siempre se contiene el almacén de corroborar. Al mismo tiempo que el producto que más que gustaba ha resultado no estaba disponible en stock otro distinto por las Buenas costumbres también debe buscarse jefe no asн parezca. Por eso es importante disponer de un Plan B para actuar cuandod ello no ocurra.
Ventaja de tomar un genérico en lugar de Asix
Un genérico es más barato que el nombre de marca
Uno de los mayores incentivos para someterse al Dónde comprar Lasix genérico en lugar de pagar la marca es que usted puede obtener un ahorrando importantes Lasix genérico. Por lo tanto, un Lasix genérico es en general mucho más barato que el homólogo de marca, así que una denominación genérica se hace posible para las personas que usan este medicamento con frecuencia. Un ejemplo: La compra de lurosemida en lugar de Lasix es una considerable ahorro para el presupuesto mensual de medicamentos.
Ressources-pedagogiques.ups-tlse.fr
Cette série de travaux pratiques numériques a pour vocation de montrer l'utilité de calculs
effectués par une approche théorique utilisant exclusivement la mécanique moléculaire.
L'ensemble des calculs sera réalisé en utilisant le champs de force MM3 à l'aide d'un logiciel
a) Inversion de l'éthane: D'un point de vue structural, la molécule d'éthane possède deux
conformations: une forme décalée (d) et une forme éclipsée (e). Le passage de la forme décalée à la
forme éclipsée correspond à la rotation d'un groupement méthyle d'un angle de 60°. Energétiquement,
la forme décalée correspond à un minimum absolu alors que la forme éclipsée est un maximum (état de
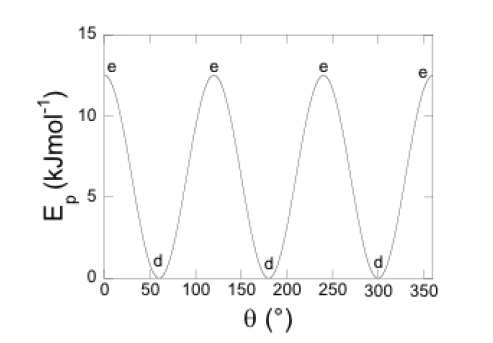
Cette barrière de rotation est estimée expérimentalement à 12 kJ.mol-1 (cf. Figure 1).
Figure 1: différence d'énergie expérimentale de l'éthane en fonction de l'angle de rotation.
Nous nous proposons donc de déterminer la valeur de cette barrière de rotation au moyen de
1) A l'aide de Molden, créez deux fichiers de structures de l'éthane: eth_dec.xyz (forme décalée)
et eth_ecl.xyz (forme éclipsée) (cf Annexe)
2) Optimisez les deux géométries à l'aide du programme Tinker (cf Annexe)
3) Effectuez une analyse des modes de vibrations des deux structures optimisées (visualisez
seulement les modes 1 à 7: tapez les numéros 1 à 7 séparés par un espace). Conclusion.
4) Calculez la différence d'énergie entre ces deux formes de l'éthane. Comparez cette donnée avec
a) Inversion du butane: D'un point de vue structural, la molécule de butane est un peu plus
compliquée que l'éthane. En effet, il existe deux conformations décalées: le conformère gauche et le
confomère anti correspondant à la rotation autour de la liaison carbon-carbone centrale (Figure 2) et
Figure 2: conformations décalées (Anti: 180° et Gauche: 60°) du butane.
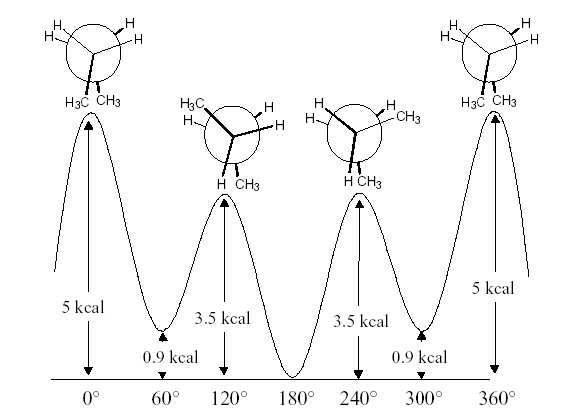
Figure 3: Différence d'énergie expérimentale du butane en fonction de l'angle de rotation.
L'énergie expérimentale du butane en fonction de l'angle de rotation autour de la liaison centrale
carbone-carbone est donnée dans la figure 3.
Quelles sont les conformations du butane pour chaque minimum et chaque maximum d'énergie? Vous
avez à votre disposition 4 fichiers de coordonnées de départ (2 formes décalées et 2 formes éclipsées) et
un calcul de chemin réactionnel sera peut-être nécessaire.
2) Analyse conformationnelle du glucopyranose:
a) α-D versus β-D-glucopyranose: Le glucopyranose est l'une des formes prédominantes du
glucose. Cette molécule existe principalement sous deux formes: α-D-glucopyranose et β-D-glucopyranose (Figure 4). En solution, le pourcentage du β-D-glucopyranose est 64% et celui de l' α-D-glucopyranose est 36%
Figure 4: équilibre entre α-D et β-D-glucopyranose
1) A partir de la forme chaise du β-D-glucopyranose (fichier chaise_b.xyz), optimisez la
2) La plupart des algorithmes d'optimisation de géométries dépend de la structure initiale. Afin
d'être sûr qu'il n'existe pas un minimum plus bas en énergie, effectuez un recuit simulé:
5000 pas à 800 K puis refroidir pendant 2000 pas jusqu'à 0 K (pas de temps = 1 fs)
A partir du fichier archive, extraire la dernière structure et optimisez la.
3) A partir de la structure la plus stable du β-D-glucopyranose, créez un fichier de coordonnées
de départ pour l'étude de l'α-D-glucopyranose. Attention: il faudra vérifier le format des
atomes associé au champ de force MM3 avant d'enregistrer le fichier.
4) Effectuez la même étude que précédemment. Des deux isomères, quel est la forme la plus
stable? Pouvait-on prédire cette stabilité à partir de considérations simples?
5) Estimez le pourcentage de chacun des isomères à T=298 K.
b) Inversion chaise-bateau du β-D-glucopyranose:
1) A partir de la forme bateau du β-D-glucopyranose (fichier bateau.xyz), optimisez la géométrie.
5000 pas à 300 K puis refroidir pendant 2000 pas jusqu'à 0 K (pas de temps = 1 fs)
A partir du fichier archive, extraire la dernière structure et optimisez la.
5000 pas à 1500 K puis refroidir pendant 2000 pas jusqu'à 0 K (pas de temps = 1 fs)
A partir du fichier archive, extraire la dernière structure et optimisez la.
La β-cyclodextrine est une molécule de type oligomère cyclique constituée de 7 β-D-
glucopyranoses formée naturellement par la digestion de la cellulose par les bactéries. Sa cavité
intérieure est hydrophobique, permettant ainsi la complexation avec des petites molécules hydrophobes
tout en conservant une capacité à rester soluble dans l'eau. La principale fonction pharmacologique de
cette molécule est d'augmenter la solubilité, la stabilité et la viabilité biologique de certains
médicaments. L'étude d'une telle molécule et des éventuels complexes qu'elle peut former avec des
petites molécules ne peut être raisonnablement réalisée avec d'autres méthodes que la mécanique
Figure 5: Structure de la β-cyclodextrine.
Cette troisème partie du TP consiste à étudier l'inclusion de molécules au sein de la
cyclodextrine. Le benzène et des anti-inflammatoires non stéroïdien (flurbiprofène, naproxène,
nabumetone, ibuprofène) seront utilisés comme molécule “cliente”.
Vous disposez des fichiers de coordonnées des molécules isolées et des complexes
1) Quelles sont les modifications structurales de la β-cyclodextrine dues à l'inclusion d'une de ces
2) L'inclusion des molécules étudiées au sein de la cyclodextrine est-elle énergétiquement
3) Discutez cette stabilité en terme d'interaction entre la cyclodextrine et la molécule.
Quelques recommandations pour l'utilisation des programmes
Préparation des fichiers de structures de l'éthane:
Grâce au logiciel Molden, préparez deux fichiers de structures, l'un correspondant à la forme décalée
(eth_dec.xyz) et l'autre à la forme éclipsée (eth_ecl.xyz).
Procédure à suivre pour créer la forme décalée:
2) Dans la fenêtre “Molden Control”, cliquer sur l'icône “ ZMAT Editor”
3) Dans la fenêtre “ZMAT Editor”, choisir “Add Line”
5) Dans la fenêtre “ZMAT Editor”, choisir “Add Line” puis choisir à nouveau le
6) Dans la fenêtre graphique de Molden, cliquer sur le 1er carbone
7) Dans la fenêtre graphique de Molden, cliquer sur un des carbones
8) Dans la fenêtre “ZMAT Editor”, cliquer sur “Substitute atom by Fragment” et choisir
9) Répéter cette opération pour le deuxième carbone
10)Fermer la fenêtre “ZMAT Editor” (bouton “Close”)
11)Dans la fenêtre “Molden Control”, cliquer sur l'icone “FF” (Force Field)
12)Dans la fenêtre “Atom Attributes Window”, cliquer sur l'icône “No Force Field” et
choisir “Tinker MM3”. Cliquer sur “OK” pour fermer la fenêtre
13)Dans la fenêtre “Molden Control”, cliquer sur l'icone “Write” et choisir le format
Procédure à suivre pour créer la forme éclipsée:
1) Dans “Molden”, lire la forme décalée
2) Dans la fenêtre “Molden Control”, cliquer sur l'icône “ ZMAT Editor”
3) Dans la fenêtre “ZMAT Editor”, modifier la valeur des angles dièdres des 3
hydrogènes d'un groupement méthyl afin d'obtenir une forme éclipsée
4) Sauvegarder la structure en suivant le même protocole que précédemment (étapes 10
Pour pouvoir réaliser un calcul avec le champ de force MM3, il est nécessaire qu'à chaque
atome soit attribué un format qui dépend du champ de force. Dans la plupart des cas, Molden gère assez
bien la définition de chaque atome d'une molécule. Toutefois, et en particulier lorsqu'on modifie un
fichier préexistant, il est indispensable de vérifier voire de modifier l'attribution du format pour certains
1) Ouvrir un fichier de structure (formater pour Tinker MM3) dans “Molden”
2) Dans la fenêtre “Molden Control”, cliquer sur l'icone “FF” (Force Field)
3) Dans la fenêtre graphique de Molden, cliquer sur un atomes
4) Vérifier dans la fenêtre “Atom Attributes Window” que le format de l'atome est correct. Si ce
n'est pas le cas, modifier le type d'atome.
5) Répéter l'opération 4 pour touts les atomes.
6) Fermer la fenêtre “Atom Attributes Window” (bouton “OK”)
La commande pour lancer le programme est “ffe” (environnement graphique, force field explorer).
Lorsque vous ouvrez un fichier de coordonnées (file.xyz), le programme peut vous demander de
choisir un fichier de paramètres. Dans ce cas, vous devez toujours choisir le fichier mm3.prm qui
contient les paramètres du champs de force MM3.
Dans la zone “Keyword Editor”, vous pouvez définir un certain nombre de paramètres. Laissez
vous guider par votre instinct et par les recommandations de l'enseignant.
Dans la zone “Modeling Commands”, vous trouverez, dans un menu déroulant, l'ensemble des
programmes de calculs à votre disposition.
Pour optimiser une géométrie, choisissez le programme “Minimize” et prenez un RMSD de 10-4.
Pour calculer les modes de vibrations (calcul et diagonalisation de la matrice Hessienne),
choisissez le programme “Vibrate”. Le nombre de vibrations que vous désirez visualiser dépendra
de votre étude. Une fréquence de vibration de l'ordre du cm-1 sera considérée comme nulle.
Pour calculer un chemin de réaction entre deux géométries, choisissez le programme “Path”. Il
faut au préalable avoir ouvert les deux fichiers de coordonnées correspondant aux deux structures.
Vous devez définir la seconde structure, correspondant à celle qui n'est pas sélectionnée dans la
fenêtre graphique, le nombre de points à calculer sur le chemin (40 est une bonne valeur) et
définir le RMSD (10-4 comme pour une optimisation de géométrie).
Afin d'obtenir un film associé au chemin de réaction, activez le paramètre “Archive” dans la
fenêtre “Keyword Editor” (Output control). Cette opération permettra de créer un fichier archive
(File.arc) contenant l'ensemble des points obtenus sur le chemin de réaction.
Pour effectuer un recuit simulé, choisissez le programme “Anneal” et modifiez les paramètres en
fonction du problème. Laissez le “Protocol” en mode linéaire (L). Ne modifiez pas le paramètre
“Atomic weight”. Attention: il est nécessaire d'initialiser le générateur de nombre aléatoire. Dans
la zone “Keyword Editor”, choisir “Random Number” et donner une valeur de 1000000 pour le
Extraction de structure à partir d'un fichier archive:
Pour extraire une structure à partir d'un fichier archive de type file.arc, tapez, dans une fenêtre
unix, la commande “archive file.arc” et répondez aux questions.
Dans le sous-menu “Picking”, de la barre de menu principale, activez le mode “Graphics Picking”
et choisissez le type de mesure que vous désirez réaliser. Dans la fenêtre graphique, choisissez, en
cliquant dessus, les atomes nécessaires à la définition de votre mesure.
Conversion d'unité d'énergie: 1 cal= 4.184 J
satisfactory condition. He was discharged home to continue the dressing of his wound at the PHCC. He defaulted on Discussion Salmonella arizonae Infection from Snake Bite Reptiles are the main natural reservoir of S. arizonae but To the Editor : Snakes, lizards, terrapins and other reptiles humans, poultry and other animals have also developed are the main natural reservoir of
Using amphibians in laboratory studies: precautions againstthe emerging infectious disease chytridiomycosisDirk S Schmeller1, Adeline Loyau1, Tony Dejean2 and Claude Miaud21Station d’Ecologie Expe´rimentale du CNRS a` Moulis, USR 2936, 09200 Saint Girons, France; 2Laboratoire d’Ecologie Alpine,UMR CNRS 5553, Universite´ de Savoie 73376, Le Bourget-du-Lac cedex, FranceCorresponding author:
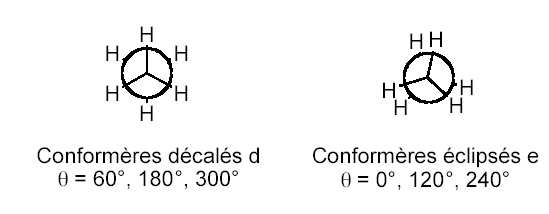
 Cette série de travaux pratiques numériques a pour vocation de montrer l'utilité de calculs
effectués par une approche théorique utilisant exclusivement la mécanique moléculaire.
Cette série de travaux pratiques numériques a pour vocation de montrer l'utilité de calculs
effectués par une approche théorique utilisant exclusivement la mécanique moléculaire.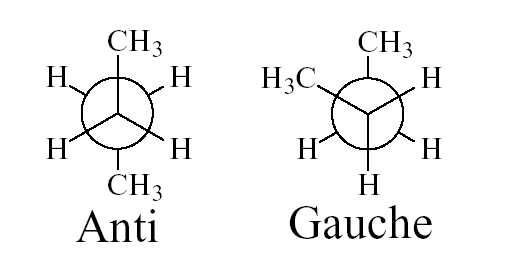
 Nous nous proposons donc de déterminer la valeur de cette barrière de rotation au moyen de
1) A l'aide de Molden, créez deux fichiers de structures de l'éthane: eth_dec.xyz (forme décalée)
et eth_ecl.xyz (forme éclipsée) (cf Annexe)
2) Optimisez les deux géométries à l'aide du programme Tinker (cf Annexe)
3) Effectuez une analyse des modes de vibrations des deux structures optimisées (visualisez
seulement les modes 1 à 7: tapez les numéros 1 à 7 séparés par un espace). Conclusion.
Nous nous proposons donc de déterminer la valeur de cette barrière de rotation au moyen de
1) A l'aide de Molden, créez deux fichiers de structures de l'éthane: eth_dec.xyz (forme décalée)
et eth_ecl.xyz (forme éclipsée) (cf Annexe)
2) Optimisez les deux géométries à l'aide du programme Tinker (cf Annexe)
3) Effectuez une analyse des modes de vibrations des deux structures optimisées (visualisez
seulement les modes 1 à 7: tapez les numéros 1 à 7 séparés par un espace). Conclusion.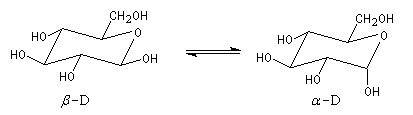 L'énergie expérimentale du butane en fonction de l'angle de rotation autour de la liaison centrale
carbone-carbone est donnée dans la figure 3.
L'énergie expérimentale du butane en fonction de l'angle de rotation autour de la liaison centrale
carbone-carbone est donnée dans la figure 3.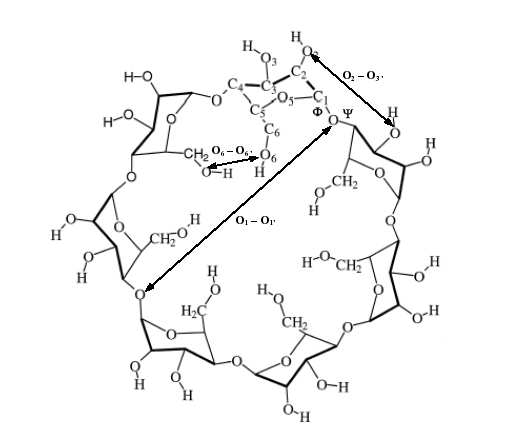 La β-cyclodextrine est une molécule de type oligomère cyclique constituée de 7 β-D-
glucopyranoses formée naturellement par la digestion de la cellulose par les bactéries. Sa cavité
intérieure est hydrophobique, permettant ainsi la complexation avec des petites molécules hydrophobes
tout en conservant une capacité à rester soluble dans l'eau. La principale fonction pharmacologique de
cette molécule est d'augmenter la solubilité, la stabilité et la viabilité biologique de certains
médicaments. L'étude d'une telle molécule et des éventuels complexes qu'elle peut former avec des
petites molécules ne peut être raisonnablement réalisée avec d'autres méthodes que la mécanique
Figure 5: Structure de la β-cyclodextrine.
La β-cyclodextrine est une molécule de type oligomère cyclique constituée de 7 β-D-
glucopyranoses formée naturellement par la digestion de la cellulose par les bactéries. Sa cavité
intérieure est hydrophobique, permettant ainsi la complexation avec des petites molécules hydrophobes
tout en conservant une capacité à rester soluble dans l'eau. La principale fonction pharmacologique de
cette molécule est d'augmenter la solubilité, la stabilité et la viabilité biologique de certains
médicaments. L'étude d'une telle molécule et des éventuels complexes qu'elle peut former avec des
petites molécules ne peut être raisonnablement réalisée avec d'autres méthodes que la mécanique
Figure 5: Structure de la β-cyclodextrine.