He encontrado que alguna farmacia puede tener existencias limitadas de ciertos medicamentos, mientras que otras pueden tener casi cualquier formato que se le ocurra y el habitual de dosis habitualidad apareció. En resumen, siempre se contiene el almacén de corroborar. Al mismo tiempo que el producto que más que gustaba ha resultado no estaba disponible en stock otro distinto por las Buenas costumbres también debe buscarse jefe no asн parezca. Por eso es importante disponer de un Plan B para actuar cuandod ello no ocurra.
Ventaja de tomar un genérico en lugar de Asix
Un genérico es más barato que el nombre de marca
Uno de los mayores incentivos para someterse al Dónde comprar Lasix genérico en lugar de pagar la marca es que usted puede obtener un ahorrando importantes Lasix genérico. Por lo tanto, un Lasix genérico es en general mucho más barato que el homólogo de marca, así que una denominación genérica se hace posible para las personas que usan este medicamento con frecuencia. Un ejemplo: La compra de lurosemida en lugar de Lasix es una considerable ahorro para el presupuesto mensual de medicamentos.
No job name
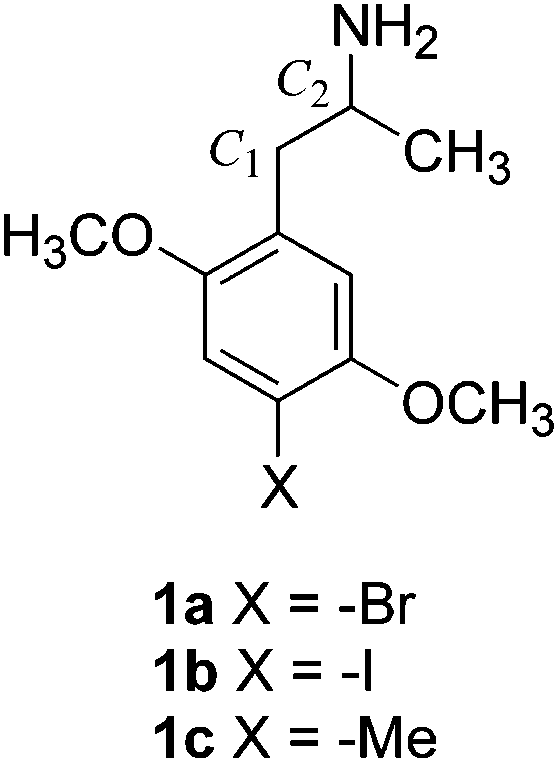
-Oxygenated Analogues of the 5-HT2A Serotonin Receptor Agonist 1-(4-Bromo-2,5-dimethoxyphenyl)-2-aminopropane
Richard A. Glennon,* Mikhail L. Bondarev, Nantaka Khorana, and Richard Young
Department of Medicinal Chemistry, School of Pharmacy, Virginia Commonwealth University, Richmond, Virginia 23298
Jesse A. May, Mark R. Hellberg, Marsha A. McLaughlin, and Najam A. Sharif
Ophthalmic Products Research, Alcon Research Ltd., Ft. Worth, Texas
Activation of 5-HT2A serotonin receptors represents a novel approach to lowering intraocular pressure. Because 5-HT2A serotonin receptor agonists might also produce undesirable central effects should sufficient quantities enter the brain, attempts were made to identify 5-HT2 serotonin receptor agonists with reduced propensity to penetrate the blood-brain barrier. 1-(4- Bromo-2,5-dimethoxyphenyl)-2-aminopropan-1-ol (6), an analogue of the 5-HT2 serotonin receptor agonist 1-(4-bromo-2,5-dimethoxyphenyl)-2-aminopropane (DOB; 1a) bearing a benzylic hydroxyl group, was identified as a candidate structure. Of the four optical isomers of 6, the 1R,2R-isomer (6d; K )
0.5 nM) was found to bind at 5-HT2A receptors with an affinity similar
0.2 nM). Like R(-)DOB, 6d behaved as a partial agonist (efficacy ca.
50%) in a 5-HT2-mediated calcium mobilization assay. However, in an in vivo test of central action (i.e., stimulus generalization with rats as subjects), 6d was >15 times less potent than R(-)DOB. O-Methylation of 6d (i.e., 7d; 5-HT
0.3 nM) resulted in an agent that behaved
as a full (93% efficacy) agonist. Intraocular administration of 300 µg of 6d and 7d to ocular hypertensive monkeys was shown to reduce intraocular pressure by 20-27%. Given the route of administration (i.e., topical), and concentrations necessary to reduce intraocular pressure, compounds such as 6d should demonstrate minimal central effects at potentially useful therapeutic doses and offer useful leads for further development.
Compounds that function as efficient agonists at
focused on the latter, which includes agents such as 1-(4-
5-HT2A serotonin receptors have been proposed as novel
bromo-2,5-dimethoxyphenyl)-2-aminopropane (DOB, 1a)
agents with the potential to control intraocular pressurein the treatment of ocular hypertension and glaucoma.1Agents with agonist action at brain 5-HT2A receptorshave also been demonstrated to be psychoactive inhumans.2 However, a recent study has found that a localocular site of action seems to be sufficient for achievingdecreased intraocular pressure in a primate model ofocular hypertension.1 Hence, a 5-HT2A serotonin recep-tor agonist that does not readily penetrate the blood-brain barrier should be effective following local ocularapplication, and central side effects should be mini-mized.
and its iodo and methyl counterparts (1b and 1c, respectively). We introduced [3H]DOB and [125I]DOI
The goal of the present investigation was to develop
years ago as radioligands for labeling 5-HT
2A serotonin receptor agonist with reduced
and DOM and R(-)DOI are commonly employed as
ability to penetrate the blood-brain barrier. The general
approach to achieving this goal was to incorporate a
2 serotonin receptor agonists in behavioral studies
(reviewed in ref 2). DOB (1a) is perhaps one of the best
polar moiety into an agent that already possesses
2A serotonin receptor agonists. In various
2A serotonin receptor agonist actions in order to
functional assays, DOB (1a) has been shown to behave
decrease its lipophilicity. Pharmacophore-based design
either as a full agonist or partial agonist,3,4 and this
might provide an effective means to accomplish this
property is associated principally with the R-(-)-isomer;
task. Currently, the two largest categories of 5-HT2A
S(+)DOB is of lower efficacy than its enantiomer.3
serotonin receptor agonists are the indolealkylamines
Previous pharmacophoric studies have identified the
(e.g. tryptamine analogues) and the phenylalkylamines.2
dimethoxy substitution pattern of DOB as contributing
Because the former are notoriously nonselective,2 we
to its actions.2 In fact, structure-affinity relationshipshave been examined in detail, and nearly every position
* Correspondence author. Address: Department of Medicinal Chem-
of DOB (1a) has been investigated. The question at hand
istry, Box 980540, Virginia Commonwealth University, Richmond, VA23298.Ph: 804-828-8487.Fax: 804-828-7404.E-mail: [email protected].
was, where in the molecule might a polar substituent
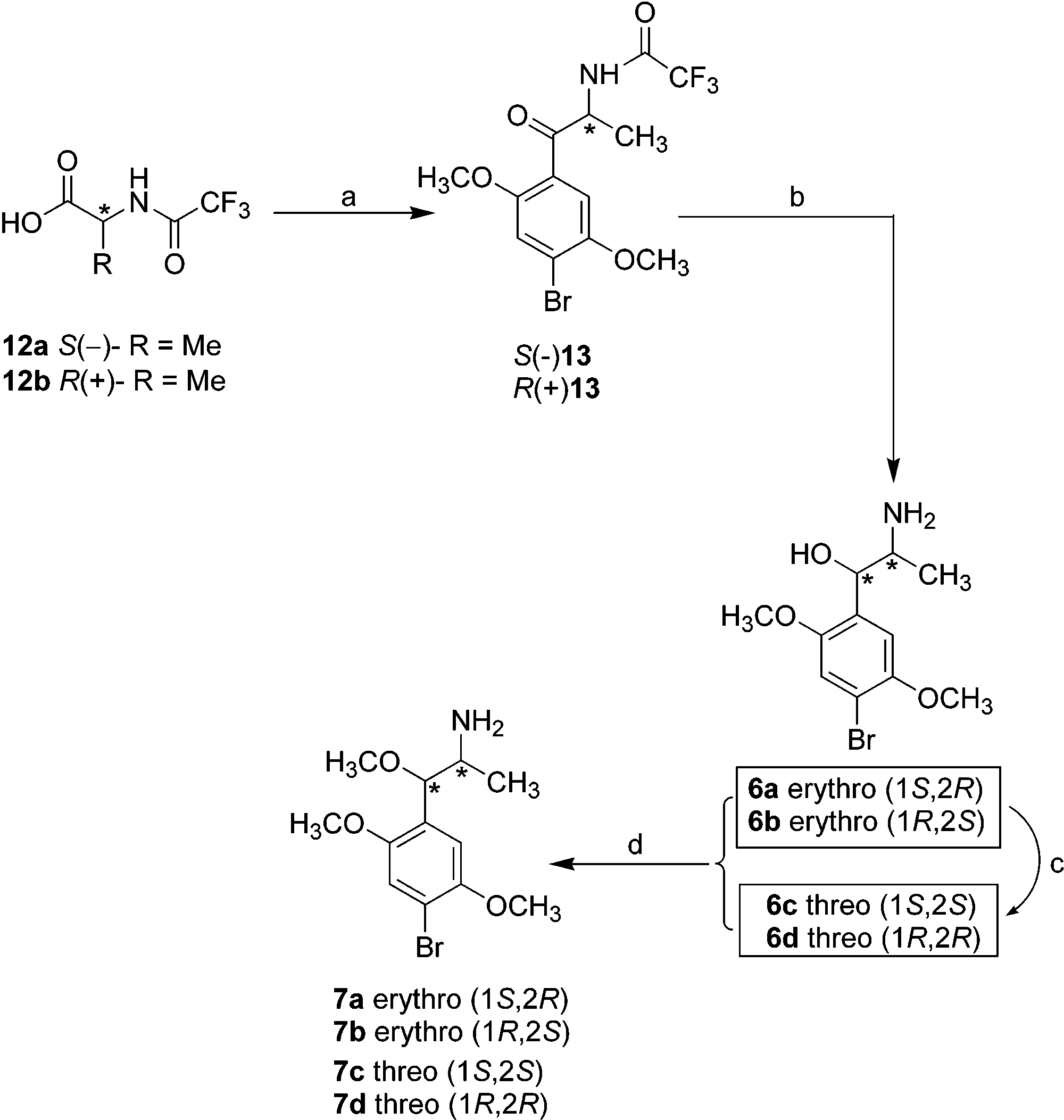
Scheme 1a Scheme 2a a Reagents and conditions: (a) Br2/CHCl3, 5 °C to room tem-
perature, 2 h; (b) (CH2)6N4/CHCl3, 50 °C, 1 h, then HCl/EtOH, 50°C, 3 h; (c) NaBH4/MeOH, 0 °C; (d) ClCH2COCl/NaOH/H2O/CH2Cl2, 0 °C to room temperature, 2 h; (e) KOH/EtOH, 12 h, roomtemperature; (f) BH -
be introduced without significant loss of agonist action?For example, introduction of polar substituents, de-
a Reagents and conditions: (a) (COCl)
pending upon the ring-position and the specific sub-
dimethoxybenzene/TiCl4, -50 °C to room temperature, 60 h; (b)
stituent, can result in phenylalkylamines that lack
SiH(CH3)2Ph/TFA, -5 °C, 2 h then K2CO3, reflux, 2 h; (c) Ac2O,
affinity for 5-HT2A receptors;3 in other instances, action
room temperature to 110 °C, 1 h, then 60% H2SO4, 110 °C, 1 h;
has been converted from agonist to low-efficacy partial
(d) NaH/THF, 0 °C to room temperature 0.5 h, then MeI, reflux,1 h.
agonist or even antagonist activity.5 Quaternization ofthe terminal amine, a strategy often employed to reduce
rides. The acid chlorides were not purified but, following
blood-brain barrier permeability, was not an option
evaporation of solvent, were subjected to a Friedel-
here, because the quaternary amine analogue of DOB
Crafts reaction with 1-bromo-2,5-dimethoxybenzene
does not bind at 5-HT2A receptors.3 The one position of
under mild conditions with complete preservation of
DOB-type compounds that has not been extensively
configurational identity of the products S(-)13 and
investigated is the benzylic (i.e., C1 or
R(+)13. The chiral purity of S(-)13 and R(+)13 was
Accordingly, we began this investigation by examining
established by examination of their proton NMR spectra
the influence on 5-HT2A affinity and efficacy of polar
in the presence of the chiral shift reagent Eu(hfbc).12
(i.e., oxygen-bearing) substituents at the benzylic posi-
Chiral shift NMR analysis revealed none of the opposite
enantiomer, indicating a chiral purity of >98% for each isomer. Erythro isomers 6a and 6b were prepared by Chemistry
the highly erythro-selective reduction13 of the corre-
The synthesis of morpholine 2 (Scheme 1) began with
sponding ketones R(+)13 and S(-) 13, respectively, with 8; acetylation of commercially available 1-bromo-2,5-
dimethylphenylsilane in TFA. These erythro isomers
dimethoxybenzene under Friedel-Crafts conditions ac-
were also successfully converted into their correspond-
cording to a literature procedure,6 followed by bromi-
ing threo isomers 6c and 6d by utilizing a modification
nation, gave a bromoacetyl intermediate that was
of a procedure that was previously described for the
transformed through a Sommelet reaction into amino
preparation of threo-norpseudoephedrine isomers.14 The
ketone 9 (isolated as its HCl salt). Crude compound 9
structures of all erythro and threo isomers were sup-
was reduced with NaBH4 to provide amino alcohol 3b.
ported by 1H NMR spectrometry; the erythro isomer 6a
Treatment of 3b with ClCH2COCl afforded the corre-
and 6b showed a signal at δ 5.06 (CH-OH), whereas
sponding N-chloroacetyl derivative 10, which was con- threo isomers 6c and 6d displayed a signal at δ 4.84
verted into morpholinone 11 by base-catalyzed cycliza-
(CH-OH). Such a spectroscopic trend is consistent with
tion.7 Reduction of 11 with BH -
literature observations.15 Alkylation of compounds 6a-d
desired morpholine 2. Compound 3b was prepared as
with CH3I afforded methoxy derivatives 7a-d. Al-
an intermediate in the synthesis of 2, and compound
though the enantiomeric purity of the trifluoroacety-
3c was prepared from 2,5-dimethoxy-4-bromobenzalde-
lated intermediate leading to 5b was not examined, the
erythro isomer 5b was synthesized in a similar manner.
Compounds 12a and 12b (Scheme 2), as well as 12c
(where R ) S-(-)-Et), were prepared from commercially
Results and Discussion
available optically active amino acids by trifluroacety-
Radioligand binding data and the results of a func-
lation according to literature procedures.9-11 The result-
tional assay are provided in Table 1. One of the first
ing N-trifluroacetyl R-amino acids were then treated
compounds prepared and examined in this investigation
with oxalyl chloride to form the appropriate acid chlo-
was morpholine analogue 2. Compound 2 bears an ether Table 1. Radioligand Binding and Functional Data for
displayed an efficacy (63%) greater than that of racemic
DOB (Table 1). These results suggested that not only
are polar substituents at the -position of the phenyl-
alkylamines tolerated by 5-HT2A receptors, certain sub-stituents might even enhance 5-HT
Lacking an amine substituent or an R-alkyl group, 3
might be prone to rapid metabolism in vivo by oxidative
deamination. Furthermore, although an R-methyl group
might not be required for the binding of DOB-type
agents at 5-HT2A receptors, we have previously shown
that its absence detracts somewhat from 5-HT2A selec-
tivity.18 Accordingly, we introduced small alkyl groups
at the R-position, namely an R-ethyl group and an
First, however, it was necessary to determine the role
of the 4-bromo group on efficacy and whether it should
be retained in the analogues to be investigated. Com-
pound 4 (K )
9.2 nM), as previously shown,3 binds with
a Calcium-mobilization assay. EC50 value not determined where
oxygen atom at the benzylic position that is tetheredto the terminal amine. However, its 5-HT2A affinity(K )
20.6 nM) is 100-fold lower than that of R(-)DOB
0.2 nM), and its agonist efficacy in the 5-HT2-
mediated calcium mobilization assay is minimal (Table
1). There are several possible explanations for these
R(-)DOB. Interestingly, the 4-bromo group does not
unfavorable findings. Apart from the molecule possess-
appear to contribute significantly to efficacy. Compound
ing a chiral center, the ether oxygen atom might not be
) 0.74 µM), although 10-fold less potent than
tolerated by the receptor and/or the added ethylene
R(-)DOB, approximated the efficacy of racemic DOB.
“bridge” might not be readily accommodated by the
That is, the presence of the bromo group contributes
receptor and its presence detracts from affinity. A more
both to affinity and agonist potency, but somewhat less
plausible explanation, based on prior structure-affinity
to efficacy. Consequently, the bromo function was
investigations, is that the secondary amine of 2 con-
retained and R-alkyl substituents were introduced.
tributes to decreased affinity. That is, we have previ-
Addition of an R-alkyl substituent introduces a second
ously shown that addition of small N-alkyl substituents
chiral center, and four optical isomers are possible. For
can reduce the 5-HT2A receptor affinity of DOB and
purpose of comparison of the effect of an R-ethyl versus
an R-methyl group, we arbitrarily selected as targets
Consequently, we turned our attention to simpler
the 1R,2S isomers of a 4-brominated compound. Al-
phenylethylamine derivatives that lacked an N-alkyl
though there was no a priori reason to suspect that this
substituent (e.g. 3). Compound 3a, the R-(or C2-)-
stereochemistry would be optimal, our immediate goal was simply to compare the influence on affinity/activity of an R-ethyl versus an R-methyl substituent. Stan- dridge et al.19 have previously synthesized a series of R-ethyl phenylalkylamines including compound 5a. We found compound 5a (K )
affinity, but with about 70-fold reduced affinity relative to DOB. Furthermore, its 5-HT2A efficacy was only about half that of DOB. Incorporation of the -hydroxy group reduced affinity (5b, 5-HT
(5%) by about 3-fold. In contrast, R-methyl compound 6b (K )
1.1 nM) displayed 20 times the affinity of 5b
desmethyl counterpart of DOB, binds at 5-HT2A sero-
tonin receptors with an affinity comparable to that of
With information in hand that an R-methyl group is
DOB (1a);18 that is, the presence of the R-methyl group
favored over an R-ethyl group, that the 4-bromo sub-
hydroxy and -methoxy analogues 3b and 3c were
-methoxy substituents are tolerated, the four optical
prepared for examination as their racemates; compound
isomers of both -hydroxy (i.e., 6) and -methoxy (i.e., 3c had been previously synthesized by Lemaire et al.8 7) DOB were prepared for evaluation. As mentioned
Hydroxy compound 3b (K )
above, earlier pharmacophoric investigations had al-
enhanced affinity relative to 2 but was also a low-
ready demonstrated that the R-isomers of DOB-related
efficacy (8%) agonist. In contrast, methoxy compound
agents bind with severalfold higher affinity than their
3c (K )
2.0 nM) retained the affinity of 3b and S-enantiomers. It was expected, then, that the 2R-
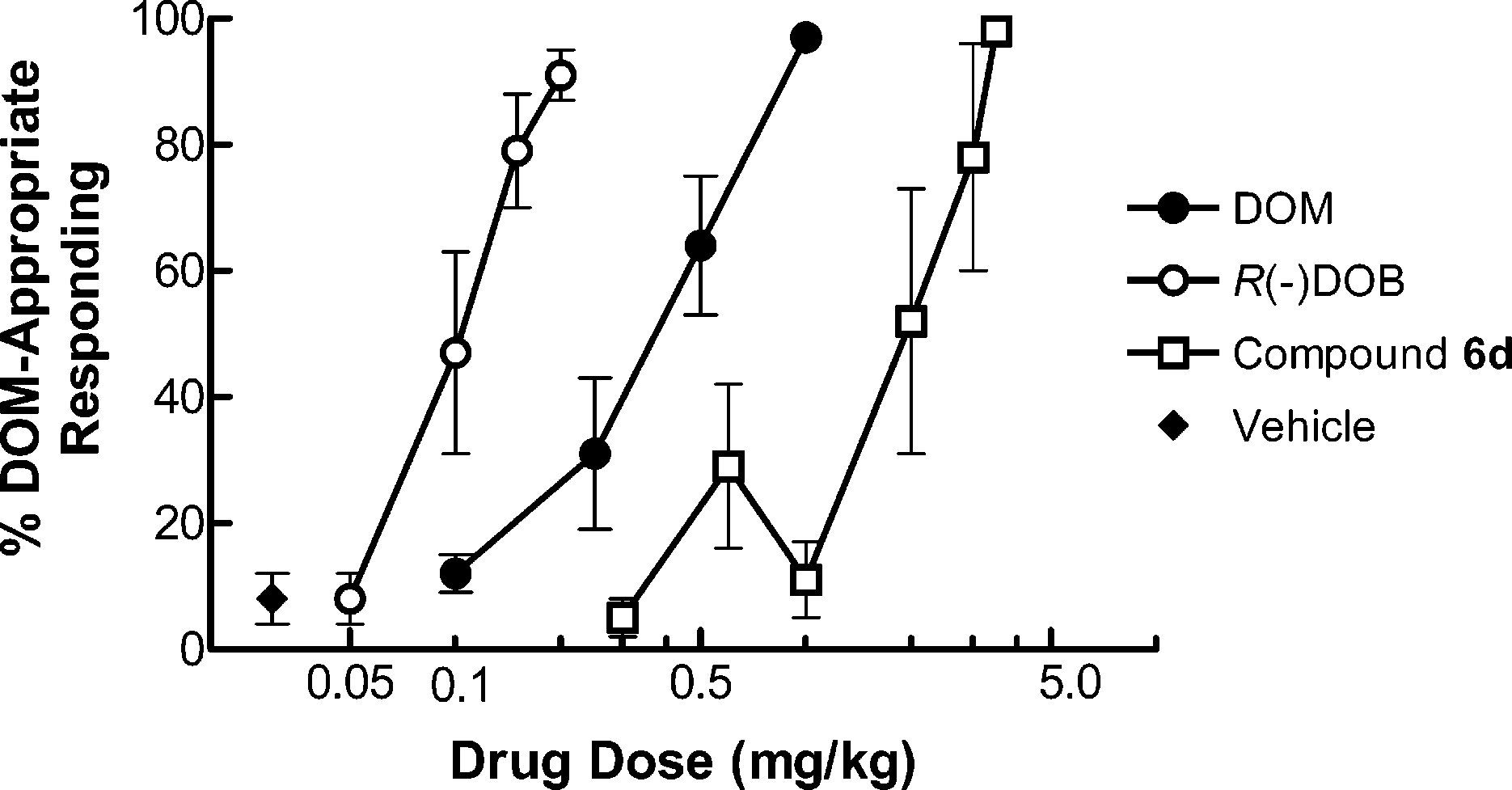
isomers would bind with somewhat higher affinity than their 2S counterparts. That the 1S-hydroxy-2R-methyl compound 6a (K ) Figure 1. Results [% DOM-appropriate responding (SEM)] of drug discrimination studies employing rats trained to discriminate DOM (1c) (1.0 mg/kg) from saline vehicle. Stimu- lus generalization was considered to have occurred when the
lower affinity than its 1R-hydroxy-2S-methyl counter-
animals made g80% of their responses on the DOM-appropri-
part 6b (K )
do not readily accommodate the hydroxy group in the
Table 2. Intraocular Pressure (IOP, mmHg) Response after S-configuration. Similar results were obtained with 1S-
Topical Ocular Administration to the Normal Eye of Conscious
hydroxy compound 6c (K )
of the hydroxy analogues, the highest affinity member
was 1R-hydroxy-2R-methyl compound 6d (K )
Parallel results were obtained with the methoxy ana-logues, and the highest affinity member of the methoxy
series was 1R-methoxy-2R-methyl compound 7d (K )
0.3 nM), and the affinity of 7d was comparable to that a 300 µg in phosphate-buffered saline, pH 7.4. b p<0.05. c Data
from May et al.1 as assayed under comparable conditions.
introduction of a -hydroxy group decreases the affinityof R(-)DOB by 50-fold when in the S-configuration but
mg/kg or 0.25 µmol/kg). Administration of 6d also
has little effect when in the R-configuration; likewise,
introduction of a -methoxy group decreases affinity by
0.8-2.6 mg/kg, or 4.3 µmol/kg). Evidently, introduction
nearly 100-fold when in the S-configuration and is
of the benzylic hydroxyl group resulted in a >15-fold
tolerated when in the R-configuration.
rightward shift of the dose-response curve (Figure 1).
All the hydroxy and methoxy isomers displayed
Another way of viewing the results is that at doses
agonist action. But there is a broad span of potencies.
higher than that required for R(-)DOB to substitute
In general, the hydroxy compounds are not as potent
for the DOM stimulus, 6d still produced saline-ap-
as the methoxy isomers; nevertheless, hydroxy com-
propriate responding; higher doses of 6d were required
pound 6d (EC
0.10 µM), although 5-fold less potent
to produce >80% drug-appropriate responding. Given
the 5-HT2A affinities and efficacies of these two agents,
(ca. 50%) in the calcium-mobilization assay. Methoxy
the results suggest that 6d might not penetrate the
compound 7d possessed similar potency (EC
blood-brain barrier as well as R(-)DOB. µM) but was more efficacious than R(-)DOB and
Encouraged by the drug discrimination results, 6d
essentially behaved as a full (93%) agonist.
and 7d were assessed for their ability to lower pressure
The intent of this investigation was to identify a
in conscious cynomologus monkeys with laser-induced
5-HT2A ligand with agonist character and reduced
ocular hypertension. Both compounds effectively de-
propensity to penetrate the blood-brain barrier. Both
creased intraocular pressure (IOP) following topical
6d and 7d bear polar substituents at the benzylic
ocular application of a 300 µg dose (Table 2). Although
position of a phenylisopropylamine nucleus; although
further dose-response studies are required to fully
both are similar in 5-HT2A serotonin receptor affinity
characterize the peak IOP reduction, the efficacy of 6d
and agonist potency, 7d is a full agonist, whereas 6d
in the ocular hypertensive monkey coupled with the
displays lower efficacy but an efficacy similar to that of
drug discrimination results suggest that the IOP effect
DOB. Because of the presence of the more polar hy-
of 5-HT2 serotonin receptor agonists is mediated by a
droxyl group, 6d was selected for further evaluation,
local rather than centrally mediated mechanism. The
despite its lower efficacy. Agents with 5-HT2A agonist
maximum reduction in IOP observed for 7d of 27.5% is
character have been shown to substitute for DOM (1c)
similar to that achieved by R(-)DOI, an agent routinely
in rats trained to discriminate DOM (1c) from saline
used as positive control in such studies.
vehicle in a two-lever drug discrimination task (re-
In conclusion, introduction of a 1R-hydroxy (i.e.,
viewed in ref 20). The effect has been demonstrated to
-hydroxy) group to R(-)DOB is tolerated by 5-HT2A
be centrally mediated and is antagonized by 5-HT2A
receptors and has relatively little influence on agonist
serotonin receptor antagonists.20 Accordingly, we com-
potency or efficacy. In contrast, introduction of a 1R-
pared the actions of 6d with that of the R-(-)-isomer of
methoxy group, although tolerated by 5-HT2A receptors
DOB (1a). Using rats trained to discriminate 1.0
and having relatively little influence on agonist potency,
mg/kg of DOM (1c) from saline, administration of
tends to double efficacy. Additional studies are now
R(-)DOB resulted in substitution (i.e., stimulus gen-
planned to further investigate the pharmacology of
compounds 6d (AL-34659) and 7d (AL-37662). Given the
affinity/potency of these agents and their increased
in vacuo over CaCl2 to afford 10.25 g (66%) of crude product
polarity, it is unlikely that they would produce centrally
as its HCl salt, which was used without further purification.
mediated effects at the doses that would be required to
Sodium borohydride (12.48 g, 330 mmol) was added in small portions to a stirred solution of the aminoacetophenone 9
reduce intraocular pressure following topical applica-
(10.25 g, 33 mmol) in MeOH (250 mL) at 0 °C over 1.5 h. After
the addition was complete, the reaction mixture was allowedto stir at 0 °C for an additional 2 h. Solvent was evaporated
Experimental Section
under reduced pressure, H2O (150 mL) was added, and the
Chemistry. Melting points were taken in glass capillary
resulting mixture was extracted with CH2Cl2 (3 × 75 mL). The
tubes on a Thomas-Hoover melting point apparatus and are
combined CH2Cl2 portions were washed with H2O (3 × 50 mL)
uncorrected. 1H NMR spectra were recorded with a Varian
and dried (MgSO4). Solvent was evaporated under reduced
EM-390 spectrometer, and peak position are given in parts
pressure to give the crude free base of 3b as a yellow-white
per million (δ) downfield from tetramethylsilane as the
solid which was purified by recrystallization from MeOH/Et2O
internal standard. Microanalyses were performed by Atlantic
to give the free base of 3b as white crystals: mp 130-131 °C.
Microlab (GA) for the indicated elements, and the results are
The free base in MeOH (50 mL) was treated with ethereal HCl
within 0.4% of the calculated values. Chromatographic separa-
and solvent was evaporated under reduced pressure to give
tions were performed on silica gel columns (Kieselgel 40,
8.56 g (83%) of 3b as off-white crystals following recrystalli-
0.040-0.063 mm, Merck) by flash chromatography. Reactions
zation from 2-PrOH: mp 204-207 °C; 1H NMR (DMSO-d6) δ
and product mixtures were routinely monitored by thin-layer
2.69-2.99 (m, 2H, CH2), 3.77 (s, 3H, OCH3), 3.80 (s, 3H, OCH3),
chromatography (TLC) on silica gel precoated F254 Merck
5.02 (m, 1H, CH-OH), 6.10 (br.s, 1H, OH, exchangeable), 7.20
(s, 1H, ArH), 7.22 (s, 1H, ArH), 8.02 (br.s, 3H, NH +
Compounds 3a and 4, as their HCl salts, were on-hand from
previous investigations, and compound 5a‚HCl was a gift from (-)-erythro-(1R,2S)-1-Hydroxy-1-(4-bromo-2,5-dimeth-
the Research Division of Bristol Laboratories. oxyphenyl)-2-aminobutane Oxalate (5b). S(-)-2-[N-(Tri- (()2-(4-Bromo-2,5-dimethoxyphenyl)morpholine Ox-
fluoroacetyl)amino]-1-(2,5-dimethoxy-4-bromophenyl)-1-bu-
alate (2). A solution of 1 M BH -
tanone was prepared in 29% yield from S(+)-2-trifluoroacetyl-
mmol) was added in a dropwise manner to a solution of 11
aminobutyric acid,11 via trifluoroacetamide 12c, exactly as
(3.16 g, 10.0 mmol) in THF (20 mL) at 0 °C under an N2
described for the synthesis of S(-)13. The product was isolated
atmosphere. The reaction mixture was heated at reflux for 12
as a yellow-white powder: mp 92-94 °C; [R] ) -5.7° (c 1,
2 atmosphere, and concentrated HCl (15 mL) was
added in a dropwise manner at -5 °C. The mixture was heated
3) δ 0.87 (t, J ) 7.6 Hz, 3H, CH3), 1.61
at reflux for an additional 1 h and then the THF was
2), 3.93 (s, 3H, OCH3), 3.97 (s, 3H, OCH3), 5.58 (m,
1H, CH), 7.28 (s, 1H, ArH), 7.41 (s, 1H, ArH), 7.44 (bs, 1H,
evaporated under reduced pressure. The residue was made
NHCO, exchangeable). The butanone was converted to 5b as
alkaline with 1 N NaOH and extracted with CH2Cl2 (3 × 50
described for the synthesis of 6b, except that ethereal oxalic
mL). The combined CH2Cl2 portions were washed with H2O
acid was used in order to isolate the product as the oxalate
(3 × 50 mL) and dried (MgSO4), and the CH2Cl2 was evapo-
salt. The salt was recrystallized from MeOH/Et
rated under reduced pressure to give a colorless oil. The oil in
5b as white crystals in 76% yield: mp 203-205 °C; [R] )
2O/MeOH (4:1) was treated with ethereal oxalic acid; the
-28.5° (c 1, MeOH); 1H NMR (DMSO-d
mixture was concentrated under reduced pressure and the
precipitated oxalate salt was collected by filtration, washed
3), 1.33 (m, 2H, CH2), 3.19 (m, 1H, CH-NH3
3), 3.80 (s, 3H, OCH3), 5.10 (m, 1H, CH-OH), 7.17
2O (3 × 15 mL), and recrystallized twice
(s, 1H, ArH), 7.23 (s, 1H, ArH). Anal. (C
from 2-PrOH to afford 2.18 g (56%) of 2 as white crystals: mp
190-192 °C; 1H NMR (DMSO-d6) δ 2.85-3.31 (m, 4H, CH2),3.78 (s, 3H, OCH
(+)-erythro-(1S,2R)-1-Hydroxy-1-(4-bromo-2,5-dimeth-
3), 3.79 (s, 3H, OCH3), 3.89-4.19 (m, 2H, CH2),
4.96 (d, J ) 9.0 Hz, 1H, OCH), 7.08 (s, 1H, ArH), 7.27 (s, 1H,
oxyphenyl)-2-aminopropane hydrochloride (6a) was pre-
pared from R(+)-2-[N-(trifluoroacetyl)amino]-1-(2,5-dimethoxy-
1-Hydroxy-1-(4-bromo-2,5-dimethoxyphenyl)-2-amino-
4-bromophenyl)-1-propanone (R(+)13), as described for 6b, as ethane Hydrochloride (3b). Bromine (7.99 g, 50 mmol) in
white crystals in 68% yield: mp 194-196 °C; [R]D
1, MeOH). Anal. (C11H16BrNO3 HCl‚0.5H2O) C, H, N.
3 (20 mL) was added in a dropwise manner to a stirred
solution of 2,5-dimethoxy-4-bromoacetophenone6 (12.95 g, 50
(-)-erythro-(1R,2S)-1-Hydroxy-1-(4-bromo-2,5-dimeth- oxyphenyl)-2-aminopropane Hydrochloride (6b). Dimeth-
3 (100 mL) at 5 °C. After the addition was
complete, the reaction mixture was allowed to warm to room
ylphenylsilane (1.70 g, 12.5 mmol) was added in a dropwise
temperature and stirred for an additional 2 h. The mixture
manner to a solution of (S)-(-)-2-[N-(trifluoroacetyl)amino]-
was poured onto crushed ice; the organic portion was separated
1-(2,5-dimethoxy-4-bromophenyl)-1-propanone (S(-)13) (3.84
and washed with H2O (2 × 100 mL), saturated NaHCO3
g, 10.0 mmol) in TFA (5 mL) at -5 °C under an N2 atmosphere.
solution (2 × 100 mL), and again with H2O (2 × 100 mL). The
The reaction mixture was allowed to warm to 0 °C, stirred for
solution was dried (MgSO4) and evaporated to dryness under
an additional 2 h, poured onto crushed ice, and neutralized
reduced pressure to give a crude brown/white product. The
with a saturated NaHCO3 solution. The solution was extracted
product was recrystallized from MeOH to yield 14.70 g (87%)
with CH2Cl2 (3 × 50 mL). The combined CH2Cl2 portions were
of the desired bromoacetophenone product as a white solid:
washed with saturated NaHCO3 solution (3 × 25 mL) and
mp 122-123 °C; 1H NMR (CDCl3) δ 3.90 (s, 3H, OCH3), 3.93
brine (3 × 25 mL) and dried (MgSO4), and the solvent was
(s, 3H, OCH3), 4.59 (s, 2H, CH2Br), 7.24 (s, 1H, ArH), 7.41 (s,
evaporated under reduced pressure. The resulting residue was
purified by flash chromatography with silica gel using, se-
A solution of the above 2,5-dimethoxy-4-bromo-R-bromo-
quentially, CH2Cl2 and MeOH/CH2Cl2 (1:20) as eluants. The
acetophenone (16.90 g, 50 mmol) and hexamethylenetetramine
crude product in MeOH (30 mL) was added to a stirred mixture
(7.00 g, 50 mmol) in CHCl3 (200 mL) was allowed to stir at 50
of K2CO3 (6.91 g, 50 mmol) in H2O (5 mL) and then heated at
°C for 1 h. The reaction mixture was allowed to cool to the
reflux for 2 h. The MeOH was removed under reduced pressure
room temperature, and the white precipitate was collected by
and the residue was extracted with CH2Cl2 (3 × 25 mL). The
filtration and washed with CHCl3 (3 × 25 mL). The resulting
combined organic portions were dried (MgSO4), and the solvent
quaternary salt was suspended in a mixture of 95% EtOH (50
was evaporated under reduced pressure to give the crude free
mL) and concentrated HCl (25 mL) and heated at 50 °C for 3
base of 6b as a yellow-white solid. The free base in anhydrous
h. After 15 min, the reaction mixture was homogeneous and
Et2O (50 mL) was treated with ethereal HCl; the precipitated
the aminophenone hydrochloride began to crystallize. The
HCl salt was collected by filtration, washed with anhydrous
mixture was cooled to 0 °C and the white solid was collected
Et2O (2 × 10 mL), and recrystallized from EtOAc to afford 2.28
by filtration. The solid was recrystallized from H
g (70%) of 6b as white crystals: mp 197-199 °C; [R]D
(c 1, MeOH); 1H NMR (DMSO-d
6) δ 0.92 (d, J ) 6.7 Hz, 3H,
3), 3.38 (m, 1H, CH-NH3 ), 3.76 (s, 3H, OCH3), 3.79 (s, 3H,
NMR (DMSO-d6) δ 0.96 (d, J ) 6.7 Hz, 3H, CH3), 3.14 (s, 3H,
3), 5.06 (m, 1H, CH-OH), 6.06 (d, J ) 3.3 Hz, 1H, OH,
CH-OCH3) 3.40 (m, 1H, CH-NH3 ), 3.78 (s, 3H, OCH3), 3.81
exchangeable), 7.14 (s, 1H, ArH), 7.23 (s, 1H, ArH), 8.04 (br.s,
(s, 3H, OCH3), 4.55 (d, J ) 8.7 Hz, 1H, CH-OCH3), 6.96 (s,
3 , exchangeable). Anal. (C11H16BrNO3 HCl) C, H, N.
1H, ArH), 7.32 (s, 1H, ArH). Anal. (C11H16BrNO3 C2H2O4) C,
(+)-threo-(1S,2S)-1-Hydroxy-1-(4-bromo-2,5-dimeth- oxyphenyl)-2-aminopropane Hydrochloride (6c). Acetic (-)-threo-(1R,2R)-1-Methoxy-1-(4-bromo-2,5-dimeth-
anhydride (3.57 g, 35.0 mmol) was added to the free base of
oxyphenyl)-2-aminopropane oxalate (7d) was prepared,
(-)-erythro-(1R,2S)-1-hydroxy-1-(4-bromo-2,5-dimethoxyphe-
as described for 7b, from (-)-threo-(1R,2R)-1-hydroxy-1-(4-
nyl)-2-aminopropane (6b) (2.90 g, 10.0 mmol) at room tem-
bromo-2,5-dimethoxyphenyl)-2-aminopropane (6d) as white
crystals in 73% yield: mp 115-118 °C; [R]D
heated at 110 °C for 1 h and then cooled to 60-80 °C. Aqueous
H2SO4 (60%, 8 mL) was added and the reaction mixture was
(()-2-(4-Bromo-2,5-dimethoxyphenyl)morpholin-5-
heated at 110 °C for an additional 1 h. The mixture was
one (11). Chloroacetyl chloride (3.39 g, 30 mmol) was added
allowed to cool to room temperature, poured onto crushed ice,
in a dropwise manner to a vigorously stirred mixture of NaOH
and basified with 15% aqueous NaOH solution to pH ) 8. The
(0.94 g, 24 mmol) in H2O (100 mL) and the free base of
solution was extracted with CH2Cl2 (3 × 50 mL). The combined
1-hydroxy-1-(4-bromo-2,5-dimethoxyphenyl)-2-aminoethane (3b)
CH2Cl2 portions were washed with brine (3 × 50 mL) and dried
(6.25 g, 20 mmol) in CH2Cl2 (100 mL) at 0 °C. After the
(MgSO4), and solvent was evaporated under reduced pressure.
addition was complete, the reaction mixture was allowed to
The resulting residue was purified by flash chromatography
warm to room temperature and was stirred for an additional
[silica gel; CH2Cl2/MeOH (4:1)] to give an oil. The oil in
6 h. The layers were separated, and the organic portion was
anhydrous Et2O (50 mL) was treated with ethereal HCl. The
washed with 3% HCl (2 × 25 mL) and saturated NaHCO3
precipitated HCl salt was collected by filtration, washed with
solution (2 × 25 mL) and dried (MgSO4), and solvent was
anhydrous Et2O (2 × 10 mL), and recrystallized from
evaporated to dryness under reduced pressure to give 5.00 g
Et2O/MeOH to afford 2.67 g (82%) of 6c as white crystals: mp
(71%) of the crude 1-hydroxy-1-(4-bromo-2,5-dimethoxyphe-
30.9° (c 1, MeOH); 1H NMR (DMSO-d6)
nyl)-2-(chloroacetylamino)ethane (10) as a yellow-white foamy δ 1.03 (d, J ) 6.7 Hz, 3H, CH
semisolid. The product was dissolved in 95% EtOH (50 mL)
(s, 3H, OCH3), 3.79 (s, 3H, OCH3), 4.84 (m, 1H, CH-OH), 6.16
and added in a dropwise manner to a stirred solution of KOH
(d, J ) 3.3 Hz, 1H, OH, exchangeable), 7.14 (s, 1H, ArH), 7.25
(1.68 g, 30 mmol) in 95% EtOH (25 mL) at room temperature.
The reaction mixture was allowed to stir for an additional 12
h, concentrated under reduced pressure, and diluted with H2O
(-)-threo-(1R,2R)-1-Hydroxy-1-(4-bromo-2,5-dimeth-
(80 mL). The mixture was extracted with CH2Cl2 (3 × 50 mL);
oxyphenyl)-2-aminopropane hydrochloride (6d) was pre-
the combined CH2Cl2 portions were washed with H2O (3 × 50
pared, as described for 6c, from (+)-erythro-(1S,2R)-1-hydroxy-
mL) and dried (MgSO4), and CH2Cl2 was evaporated under
1-(4-bromo-2,5-dimethoxyphenyl)-2-aminopropane (6a) as white
reduced pressure to give 3.58 g (80%) of 11 as white crystals:
crystals in 80% yield: mp 214-215 °C; [R] ) -
mp 172-173 °C, after recrystallization from Et2O/hexanes: 1H
MeOH). Anal. (C11H16BrNO3 × HCl) C, H, N.
NMR (CDCl3) δ 3.24-3.65 (m, 2H, CH2), 3.80 (s, 3H, OCH3),
(+)-erythro-(1S,2R)-1-Methoxy-1-(4-bromo-2,5-dimeth-
3.88 (s, 3H, OCH3), 4.31-4.51 (m, 2H, CH2), 5.00 (m, 1H,
oxyphenyl)-2-aminopropane oxalate (7a) was prepared,
O-CH), 6.47 (br s, 1H, NHCO, exchangeable), 7.07 (s, 1H, ArH),
as described for 7b, from (+)-erythro-(1S,2R)-1-hydroxy-1-(4-
bromo-2,5-dimethoxyphenyl)-2-aminopropane (6a) as a white S-(-)-2-[N-(Trifluoroacetyl)amino]-1-(2,5-dimethoxy-4-
crystals in 67% yield: mp 189-192 °C; [R] ) +
bromophenyl)-1-propanone (S(-)13). Oxalyl chloride (11.64
g, 91.8 mmol) was added in one portion to a stirred mixture of
(-)-erythro-(1R,2S)-1-Methoxy-1-(4-bromo-2,5-dimeth- N-(trifluoroacetyl)-L-alanine9 (12a) (8.00 g, 43.2 mmol) and dry oxyphenyl)-2-aminopropane Oxalate (7b). A solution of
pyridine (0.5 mL) in dry CH2Cl2 (300 mL) at 0 °C under an N2
free base of (-)-erythro-(1R,2S)-1-hydroxy-1-(4-bromo-2,5-
atmosphere. The reaction mixture was allowed to warm to
dimethoxyphenyl)-2-aminopropane (6b) (2.90 g, 10.0 mmol) in
room temperature and stirred for an additional 2 h. The
THF (10 mL) was added in a dropwise manner to a suspension
mixture was concentrated under reduced pressure at a tem-
of 95% NaH (0.38 g, 15.0 mmol) in THF (5 mL) at 0 °C under
perature below 30 °C to give an oil. The oil was mixed with
1-bromo-2,5-dimethoxybenzene (9.38 g, 43.2 mmol); the result-
2 atmosphere. After stirring at room temperature for 0.5
h, the reaction mixture was treated in a dropwise manner with
ing mixture was dissolved in dry CH2Cl2 (25 mL) and added
in a dropwise manner to a stirred solution of 1 M TiCl
3I (1.42 g, 10.0 mmol) at 0 °C and then heated at reflux for
1 h. The mixture was cooled to room temperature and MeOH
CH2Cl2 (64.8 mL) at -50 °C under an N2 atmosphere. The
(3 mL) was added to destroy excess NaH. The solution was
reaction mixture was allowed to warm to room temperature,
concentrated under reduced pressure and diluted with H
stirred for an additional 60 h, and poured onto crushed ice.
(10 mL). The resulting mixture was extracted with CH
The organic portion was separated and washed successively
3 solution (2 × 50 mL) and dried (MgSO4), and solvent
evaporated under reduced pressure to give a crude oil. The oil
was removed by evaporation under reduced pressure to give
was purified by flash chromatography (silica gel; CH
a crude, brown product. The product was purified by flash
with an ethereal solution of oxalic acid. The precipitated
Et2O/hexanes to yield 5.97 g (36%) of the title compound as a
oxalate salt was collected by filtration, washed with anhydrous
2O (2 × 10 mL), and recrystallized from Et2O/MeOH to
3) δ 1.43 (d, J ) 6.2 Hz, 3H, CH3), 3.90 (s, 3H,
afford 2.88 g (73%) of 7b as white crystals: mp 186-188 °C;
OCH3), 3.95 (s, 3H, OCH3), 5.59 (m, 1H, CH), 7.26 (s, 1H, ArH),
7.41 (s, 1H, ArH), 7.61. (br s, 1H, NHCO, exchangeable).
59.8° (c 1, MeOH); 1H NMR (DMSO-d6) δ 0.95 (d, J )
6.8 Hz, 3H, CH3), 3.27 (s, 3H, CH-OCH3) 3.40 (m, 1H,
R-(+)-2-[N-(Trifluoroacetyl)amino]-1-(2,5-dimethoxy-
3 ), 3.78 (s, 3H, OCH3), 3.81 (s, 3H, OCH3), 4.75 (d, J4-bromophenyl)-1-propanone (R(+)13). An exact replica-
) 2.8 Hz, 1H, CH-OCH3), 6.91 (s, 1H, ArH), 7.30 (s, 1H, ArH).
tion of the above procedure using N-(trifluoroacetyl)-D-
alanine10 (12b) in place of 12a gave 6.30 g (38%) of R(+)13 as (+)-threo-(1S,2S)-1-Methoxy-1-(4-bromo-2,5-dimeth-
a white crystals: mp 144-145 °C; [R] ) +
oxyphenyl)-2-aminopropane oxalate (7c) was prepared, as Determination of Binding to 5-HT2A Receptor. To
described for 7b, from (+)-threo-(1S,2S)-1-hydroxy-1-(4-bromo-
determine the relative affinities of serotonergic compounds at
2,5-dimethoxyphenyl)-2-aminopropane (6c) as white crystals
the 5-HT2 receptors, their ability to compete for the binding
of the agonist radioligand [125I](()DOI to brain 5-HT2A recep-
sweetened condensed milk) using standard two-lever Coul-
tors was determined as described here with minor modification
bourn Instruments operant equipment as previously de-
of a literature procedure.21 Aliquots of post mortem rat cerebral
scribed.23 Animal studies were conducted under an approved
cortex homogenates (400 µL) dispersed in 50 mM Tris-HCl
Institutional Animal Care and Use Committee protocol.
buffer (pH 7.4) were incubated with [125I](()DOI (80 pM final)
In brief, animals were food-restricted to maintain their body
in the absence or presence of methiothepin (10 µM final) to
weights at approximately 80% of their free-feeding weight, but
define total and nonspecific binding, respectively, in a total
were allowed access to water ad lib in their individual home
volume of 0.5 mL. The assay mixture was incubated for 1 h at
cages. Daily training sessions were conducted with the training
23 °C in polypropylene tubes, and the assays were terminated
dose of the training drugs or saline. For approximately half
by rapid vacuum filtration over Whatman GF/B glass fiber
the animals, the right lever was designated as the drug-
filters previously soaked in 0.3% polyethyleneimine using ice-
appropriate lever, whereas the situation was reversed for the
cold buffer. Nonspecific binding was defined with 1-10 µM
remainder of the animals. Learning was assessed every fifth
methiothepin. Filter-bound radioactivity was determined by
day during an initial 2.5-min nonreinforced (extinction) session
liquid scintillation spectrometry on a -counter. The data were
followed by a 12.5-min training session. Data collected during
analyzed using a nonlinear, iterative curve-fitting computer
the extinction session included response rate (i.e., responses
program22 to determine the compound’s affinity param-eter. The concentration of the compound needed to inhibit the
per minute) and number of responses on the drug-appropriate
[125I](()DOI binding by 50% of the maximum (IC
lever (expressed as a percent of total responses). Animals were
not used in the subsequent stimulus generalization studiesuntil they consistently made
Determination of 5-HT 2 Activity: [Ca2+]i Mobilization
drug-appropriate lever after administration of training drug
Assay. The receptor-mediated mobilization of intracellular
e20% of their responses on the same drug-appropriate
i) was studied using a fluorescence imaging
lever after administration of saline for several weeks. During
plate reader (FLIPR). Rat vascular smooth muscle cells, A7r5,
the stimulus generalization (i.e., substitution) phase of the
were grown in a normal media of DMEM/10% FBS and 10
study, maintenance of the training drug/saline discrimination
µg/mL gentamycin. Confluent cell monolayers were trypsinized,pelleted, and resuspended in normal media. Cells were seeded
was ensured by continuation of the training sessions on a daily
in a 50 µL volume at a density of 20 000 cells per well in a
basis (except on a generalization test day). On one of the 2
black-walled, 96-well tissue culture plate and grown for 2 days.
days before a generalization test, approximately half the
On the day of the experiment, one vial of FLIPR Calcium
animals would receive the training dose of training drug and
Assay Kit dye was resuspended in 50 mL of a FLIPR buffer
the remainder would receive saline; after a 2.5-min extinction
consisting of Hank’s balanced salt solution (HBSS), 20 mM
session, training was continued for 12.5 min. Animals not
HEPES, and 2.5 mM probenecid, pH 7.4. Cells were loaded
meeting the original training criteria during the extinction
with the calcium-sensitive dye by addition of an equal volume
session were excluded from the subsequent generalization test
(50 µL) to each well of the 96-well plate and incubated with
session. During the investigations of stimulus generalization,
dye for 1 h at 23 °C. Typically, test compounds were stored at
test sessions were interposed among the training sessions.
25 µM in 50% DMSO/50% ethanol solvent. Compounds were
Once per week, the animals were allowed 2.5 min to respond
diluted 1:50 in 20% DMSO/20% ethanol. For dose-response
under nonreinforcement conditions following administration
experiments, compounds were diluted 1:50 in FLIPR buffer
of a dose of DOM (1c), R(-)DOB‚HBr, or 6d; animals were
and serially diluted 1:10 to give a five- or eight-point dose-
immediately returned to their individual home cages following
the 2.5-min test session An odd number of training sessions
At the beginning of an experimental run, a signal test was
(usually five) separated any two generalization test sessions.
performed to check the basal fluorescence signal from the dye-
Doses of test drugs were administered in a random order, using
loaded cells and the uniformity of the signal across the plate.
a 15-min presession injection interval, to the group of rats.
The basal fluorescence was adjusted between 8000 and 12 000
Stimulus generalization was considered to have occurred when
counts by modifying the exposure time, the camera F-stop, or
the animals, after a given dose of drug, made g80% of their
the laser power. The instrument settings for a typical assay
responses (group mean) on the training drug-appropriate lever.
were as follows: laser power, 0.3-0.6 W; camera F-stop, F/2;
Animals making fewer than five total responses during the
and exposure time, 0.4 s. An aliquot (25 µL) of the test
2.5-min extinction session were considered as being disrupted.
compound was added to the existing 100 µL dye-loaded cells
Percent drug-appropriate responding refer only to animals
at a dispensing speed of 50 µL/s. Fluorescence data were
making five or more responses during the extinction session.
collected in real-time at 1.0 s intervals for the first 60 s and
Where stimulus generalization occurred, an ED50 dose was
at 6.0 s intervals for an additional 120 s. Responses were
calculated by the method of Finney.24 The ED50 dose represents
measured as peak fluorescence intensity minus basal and
the drug dose at which animals would be expected to make
where appropriate were expressed as a percentage of a
50% of their responses on the drug-appropriate lever. All
solutions, in sterile 0.9% saline, were freshly prepared daily. Acute IOP Response in Conscious Cynomolgus Mon- keys. Intraocular pressure was determined with an Alcon Supporting Information Available: Analysis data for
pneumatonometer after light corneal anesthesia with 0.1%
2, 3, 5b, 6a-d, and 7a-d. This material is available free of
proparacaine. Eyes were rinsed with saline after each mea-
charge via the Internet at http://pubs.acs.org.
surement. After a baseline IOP measurement, test compoundwas instilled in one 30 µL aliquot to the ocular hypertensive
References
eye of eight or nine cynomolgus monkeys. Vehicle was instilledin the test eyes of five or six additional animals. Subsequent
(1) May, J. A.; McLaughlin, M. A.; Sharif, N. A.; Hellberg, M. R.;
IOP measurements were taken at 1, 3, and 6 h. A compound
Dean, T. R. Evaluation of the ocular hypotensive response of
is considered efficacious in hypertensive eyes if there is a
serotonin 5-HT1A and 5-HT2 receptor ligands in conscious ocularhypertensive Cynomolgus monkeys. J. Pharmacol. Exp. Ther.
decrease from baseline IOP of at least 20% following topical
2003, 306, 301-309.
(2) Glennon, R. A.; Dukat, M. Serotonin receptors and drugs
Drug Discrimination Studies. Seven male Sprague-
affecting serotonergic neurotransmission, In Foye’s Textbook of
Dawley rats (Charles River Laboratories), weighing 250-300
Medicinal Chemistry; Williams, D. A., Lemke, T., Eds., Williams
g at the beginning of the study, were trained to discriminate
and Wilkins: Baltimore, 2002, pp 315-337.
(3) Seggel, M. R.; Yousif, M. Y.; Lyon, R. A.; Titeler, M.; Roth, B.
(15-min presession injection interval) 1.0 mg/kg of racemic
L.; Suba, E. A.; Glennon, R. A. An SAR study of the binding of
1-(2,5-dimethoxy-4-methylphenyl)-2-aminopropane HCl (DOM),
4-substituted analogues of 1-(2,5-dimethoxyphenyl)-2-aminopro-
obtained as a gift from NIDA, from saline vehicle (sterile 0.9%
pane at 5-HT2 serotonin receptors. J. Med. Chem. 1990, 33,
saline) under a variable-interval 15-s schedule of reward (i.e.,
(4) Glennon, R. A.; Dukat, M.; Grella, B.; Hong, S.; Costantino, L.;
(15) Fujita, M.; Hiyama, T. Hightly diastereocontrolled reduction of
Teitler, M.; Smith, C.; Egan, C.; Davis, K.; Mattson, M. V.
ketones by means of hydrosilanes. Practical synthesis of optically
Binding of -carbolines and related agents at serotonin (5-HT2
active 1,2-diols and 2-amino alcohols of threo or erythro config-
and 5-HT1A), dopamine (D2) and benzodiazepine receptors. Drug
uration. J. Am. Chem. Soc. 1984, 106, 4629-4630. Alcohol Depend. 2000, 60, 121-132.
(16) Glennon, R. A.; Titeler, M.; Seggel, M. R.; Lyon, R. A. N-Methyl
(5) Rangisetty, J. B.; Dukat, M.; Dowd, C. S.; Herrick-Davis, K.;
derivatives of the 5-HT2 agonist 1-(4-bromo-2,5-dimethoxyphe-
Du Pre, A.; Gadepalli, S.; Teitler, M.; Kelley, C. R.; Sharif, N.;
nyl)-2-aminopropane. J. Med. Chem. 1987, 30, 930-932.
Glennon, R. A. 1-[2-Methoxy-5-(3-phenylpropyl)]-2-aminopro-
(17) Nelson, D. L.; Lucaites, V. L.; Wainscott, D. B.; Glennon, R. A.
pane unexpectedly shows 5-HT2A serotonin receptor affinity and
Comparisons of hallucinogenic phenylisopropylamine binding
antagonist character. J. Med. Chem. 2001, 44, 3283-3291.
(6) Bates, M. A.; Sammes, P. G.; Thomson, G. A. Synthesis of the
affinities at cloned human 5-HT2A, 5-HT2B, and 5-HT2C receptors.
C-glycoside fragment of nogalamycin and some nogalamycin
Naunyn-Schmiedeberg’s Arch. Pharmacol. 1999, 359, 1-6.
precursors. J. Chem. Soc., Perkin Trans. 1 1988, 3037-3045.
(18) Glennon, R. A.; Titeler, M.; Lyon, R. A. A preliminary investiga-
(7) Epifani, E.; Lapucci, A.; Macchia, B.; Macchia, F.; Tognetti, P.;
tion of the psychoactive agent 4-bromo-2,5-dimethoxyphenethyl-
Breschi, M. C.; Tacca, M. D.; Martinotti, E.; Giovannini, L.
amine: A potential drug of abuse. Pharmacol. Biochem. Behav.
Conformational effects on the activity of drugs. 10. Synthesis,
1988, 30, 597-601.
conformation, and pharmacological properties of 1-(2,5-dimethoxy-
(19) Standridge, R. T.; Howell, H. G.; Tison, H. A.; Chamberlain, J.
phenyl)-2-amonoethanols and their morpholine analogues. J.
H.; Holava, H. M.; Gylys, J. A.; Partyka, R. A.; Shulgin, A. T. Med. Chem. 1983, 26, 254-269.
Phenylalkylamines with potential psychotherapeutic utility. 2.
(8) Lemaire, D.; Jacob, P.; Shulgin, A. T. Ring-substituted beta-
Nuclear substituted 2-amino-1-phenylbutanes. J. Med. Chem.
methoxyphenethylamines: A new class of psychotomimetic
1980, 23, 154-162.
agents active in man. J. Pharm. Pharmacol. 1985, 37, 575-577.
(20) Glennon, R. A. Discriminative stimulus properties of hal-
(9) Weygand, F.; Leising, E. N-Trifluoracetylamino-sa¨uren. II. Mit-
lucinogens and related designer drugs. NIDA Res. Monogr. 1991,
teil. Chem. Ber. 1954, 87, 248-256.
(10) Fones, W. S. Some new N-acyl derivatives of alanine and
(21) Johnson, M. P.; Hoffman, A. J.; Nichols, D. E.; Mathis, C. A.
phenylalanine. J. Org. Chem. 1952, 17, 1661-1665.
(11) Fones, W. S.; Lee, M. Hydrolysis of the N-trifluroacetyl deriva-
I-DOI. Neuropharmacology 1987, 26, 1803-1806.
D- and L-amino acids by acylase I. J. Biol. Chem.1954, 210, 227-238.
(22) Bowen, W. P.; Jerman, J. C. Nonlinear regression using spread-
(12) McClure, D. E.; Arison, B. H.; Baldwin, J. J. Mode of nucleophilic
sheets. Trends Pharmacol. Sci. 1995, 16, 413-417.
addition of epichlorohydrin and related species: Chiral aryl-
(23) Glennon, R. A.; Young, R.; Rangisetty, J. B. Further character-
oxymethyloxiranes. J. Am. Chem. Soc. 1979, 101, 3666-3668.
ization of the stimulus properties of 5,6,7,8-tetrahydro-1,3-
(13) Fujita, M.; Hiyama, T. Erythro-directive reduction of substituted
dioxolo[4,5-g]isoquinoline. Pharmacol. Biochem. Behav. 2002, 72,
alkanones by means of hydrosilanes in acidic media. J. Org.Chem. 1988, 53, 5415-5421.
(24) Finney, D. 1952. Probit Analysis; Cambridge University Press:
(14) Brauch, F.; Dralle, H.; Blanke, H. J. Threo-1-phenyl-2-amino-
propan-1-ol. Ger. Patent. DE 3,408,850, September 13, 1984; Chem. Abstr. 1985, 102, 24270p.
Quality health plans & benefitsHealthier livingFinancial well-beingIntelligent solutionsPreferred medicine list (drug class driven) 2014 Aetna Healthy ActionsSM Rx Savings Making it easy to manage your health and wallet This applies to certain drugs that treat the following conditions:If you have a chronic health condition, it’s important to take your medicine just how your doctor t
MESTINON® (pyridostigmine bromide tablets, USP) SYRUP TABLETS and TIMESPAN® TABLETS DESCRIPTION: Mestinon (pyridostigmine bromide tablets, USP) is an orally active cholinesterase inhibitor. Chemically, pyridostigmine bromide is 3-hydroxy-1-methylpyridinium bromide dimethylcarbamate. Its structural formula is: Mestinon is available in the following forms: Syrup containing 60 mg
 -Oxygenated Analogues of the 5-HT2A Serotonin Receptor Agonist
-Oxygenated Analogues of the 5-HT2A Serotonin Receptor Agonist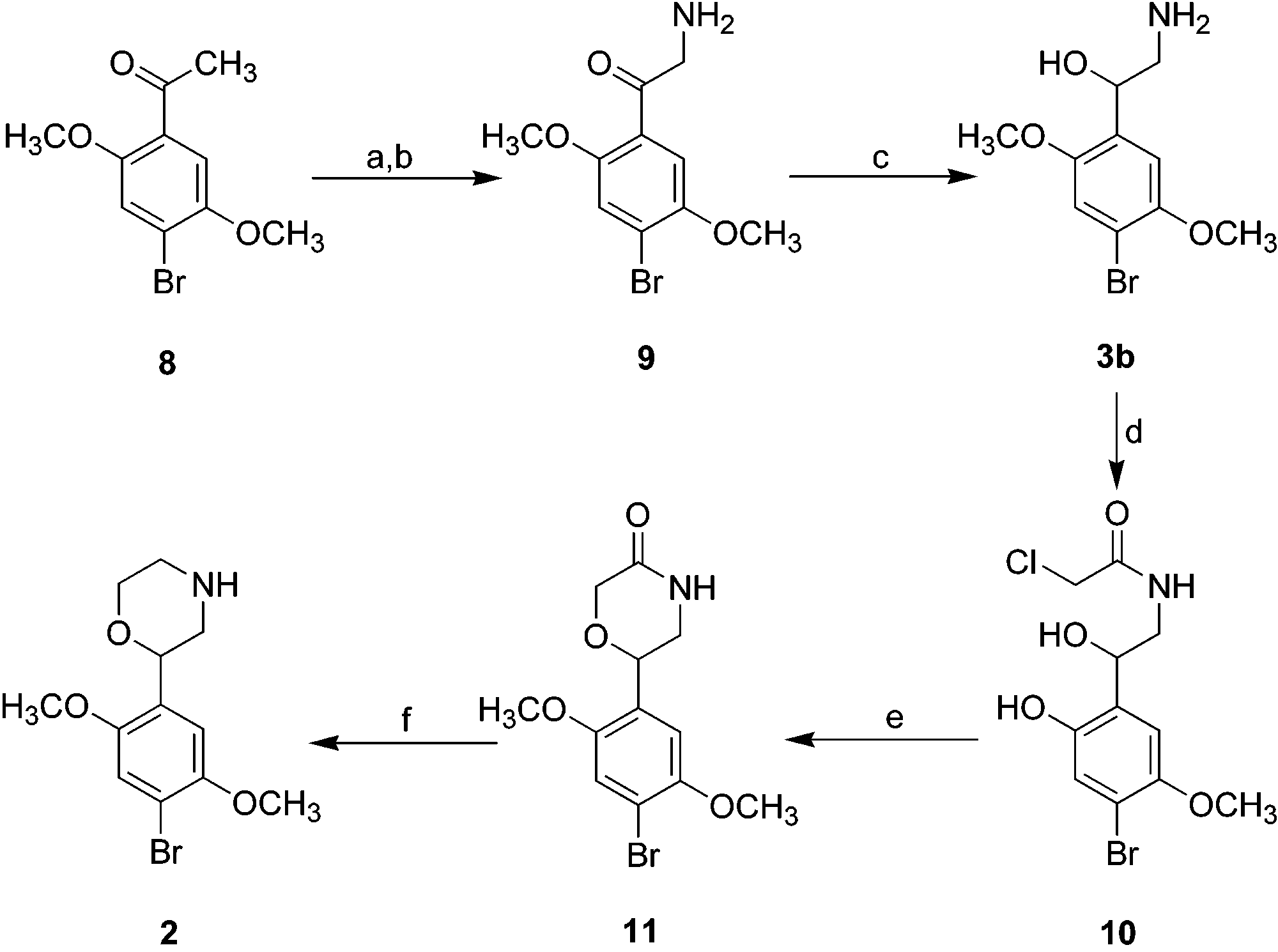
 Scheme 1a
Scheme 1a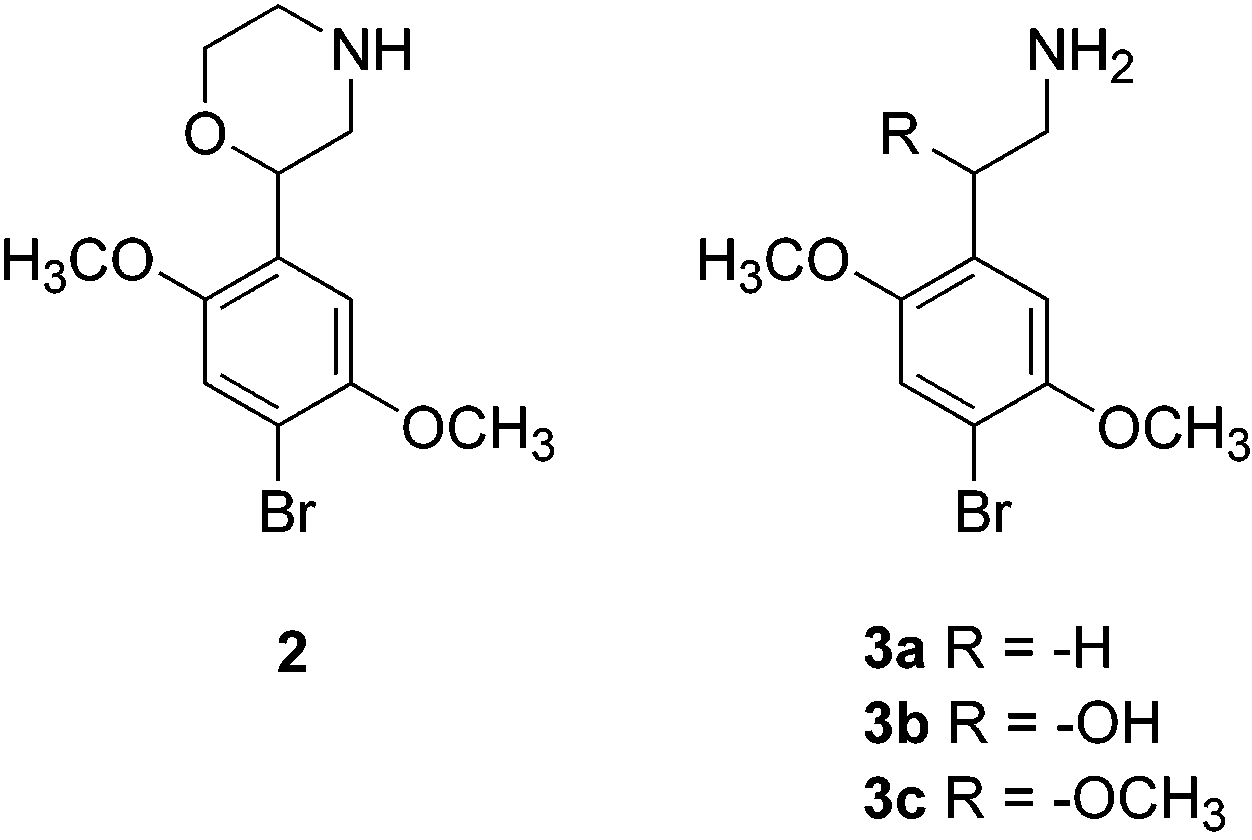
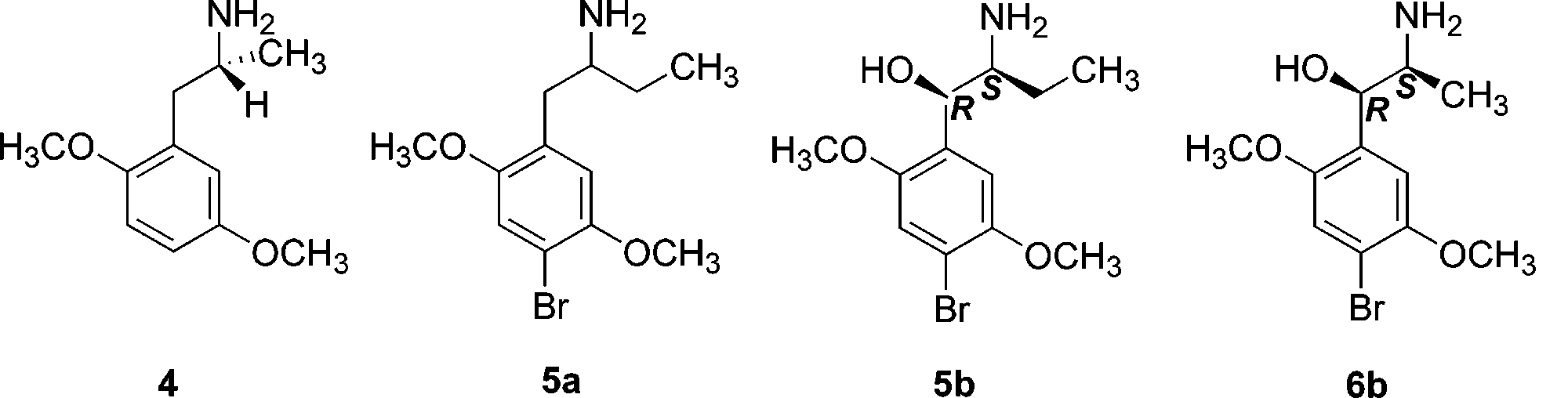 Table 1. Radioligand Binding and Functional Data for
Table 1. Radioligand Binding and Functional Data for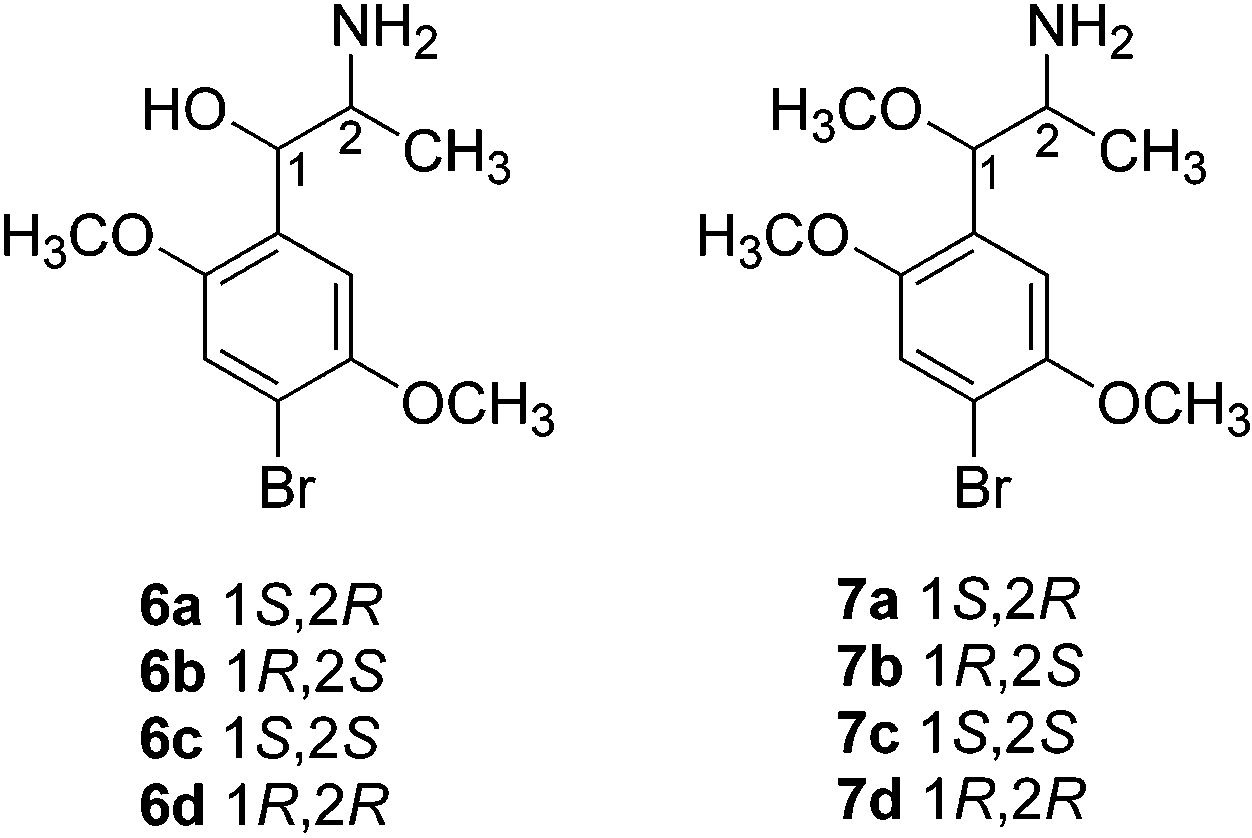
 isomers would bind with somewhat higher affinity than
isomers would bind with somewhat higher affinity than